飼料及び飼料添加物の成分規格等に関する省令
昭和51年 7月24日 農林省令第 35号
最終改正 令和 8年 5月26日 農林水産省令第 42号
別表第1(第1条関係)
1 飼料一般の成分規格並びに製造、使用及び保存の方法及び表示の基準
(1) 飼料一般の成分規格
ア 飼料は、抗菌性物質(飼料添加物として指定されたものを除く。)を含んではならない。
イ 次の表の対象飼料の欄に掲げる飼料及びうずら(産卵中のものは除く。)を対象とする飼料以外の飼料は、同表に掲げる飼料添加物を含んではならない。
ウ 次の表に掲げる対象飼料が含むことができる飼料添加物の量は、同表に掲げるとおりとする。
| 対象飼料 | 鶏(ブロイラーを除く。)用 | ブロイラー用 | 豚用 | 牛用 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 飼料添加物名 | 単位 | 幼すう用・中すう用 | 前期用 | 後期用 | ほ乳期用 | 子豚期用 | ほ乳期用 | 幼齢期用 | 肥育期用 | |
| アビラマイシン | g力価 | 2.5~10 | 2.5~10 | 2.5~10 | 10~40 | 5~40 | ||||
| エンラマイシン | g力価 | 1~10 | 1~10 | 1~10 | 2.5~20 | 2.5~20 | ||||
| サリノマイシンナトリウム | g力価 | 50 | 50 | 50 | 15 | 15 | ||||
| センデュラマイシンナトリウム | g力価 | 25 | 25 | 25 | ||||||
| ナラシン | g力価 | 80 | 80 | 80 | ||||||
| ノシヘプタイド | g力価 | 2.5~10 | 2.5~10 | 2.5~10 | 2.5~20 | 2.5~20 | ||||
| ビコザマイシン | g力価 | 5~20 | 5~20 | 5~20 | 5~20 | 5~20 | ||||
| フラボフォスフォリポール | g力価 | 1~5 | 1~5 | 1~5 | 2~10 | 2.5~5 | ||||
| モネンシンナトリウム | g力価 | 80 | 80 | 80 | 30 | 30 | 30 | |||
| ラサロシドナトリウム | g力価 | 75 | 75 | 75 | 33 | |||||
| アンプロリウム・エトパベート | g | アンプロリウム | 40~250 | 40~250 | 40~250 | |||||
| エトパベート | 2.56~16 | 2.56~16 | 2.56~16 | |||||||
| アンプロリウム・エトパベート・スルファキノキサリン | g | アンプロリウム | 100 | 100 | 100 | |||||
| エトパベート | 5 | 5 | 5 | |||||||
| スルファキノキサリン | 60 | 60 | 60 | |||||||
| クエン酸モランテル | g | 30 | 30 | |||||||
| ナイカルバジン | g | 100 | ||||||||
1 対象飼料とは、次のものをいう。
| 鶏(ブロイラーを除く。)用 | 幼すう用 | ふ化後おおむね4週間以内の鶏用飼料 |
| 中すう用 | ふ化後おおむね4週間を超え10週間以内の鶏用飼料 | |
| ブロイラー用 | 前期用 | ふ化後おおむね3週間以内のブロイラー用飼料 |
| 後期用 | ふ化後おおむね3週間を超え食用として屠殺する前7日までのブロイラー用飼料 | |
| 豚用 | ほ乳期用 | 体重がおおむね30kg以内の豚用飼料 |
| 子豚期用 | 体重がおおむね30kgを超え70kg以内の豚(種豚育成中(体重がおおむね60kgを超え120kg以内のものに限る。以下同じ。)のものを除く。)用飼料 | |
| 牛用 | ほ乳期用 | 生後おおむね3月以内の牛用飼料(モネンシンナトリウムを含むものにあつては、主として離乳後の牛の育成の用に供する配合飼料であつて、脱脂粉乳を主原料とするもの以外のものに限る。) |
| 幼齢期用 | 生後おおむね3月を超え6月以内の牛用飼料 | |
| 肥育期用 | 生後おおむね6月を超えた肥育牛(搾乳中のものを除く。)用飼料 |
2 対象飼料が含むことができる飼料添加物の量は、飼料1トン当たりの有効成分量である。
エ ギ酸(ギ酸カルシウム及び二ギ酸カリウム中に含まれるものを除く。)の飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、ギ酸として0.5%以下でなければならない。
オ プロピオン酸、プロピオン酸カルシウム及びプロピオン酸ナトリウムの飼料中の含有量は、サイレージ(牧草等(乾燥して水分含量を低下させたものを含む。)をサイロ又は適当な容器に詰め、乳酸発酵させて調製する飼料をいう。)にあっては、プロピオン酸として1.0%以下、それ以外の飼料(飼料を製造するための原料又は材料を除く。)にあっては、プロピオン酸として0.3%以下でなければならない。
カ エトキシキン、ジブチルヒドロキシトルエン及びブチルヒドロキシアニソールの飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、それぞれの有効成分の合計量で飼料1トン当たり150g以下でなければならない。
キ(ア) 魚類及び甲殻類を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料は、飼料添加物であるアスタキサンチンを含んではならない。
(イ) 飼料添加物であるアスタキサンチンの飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、魚類を対象とする飼料にあつては飼料1トン当たり100g以下、甲殻類を対象とする飼料にあつては飼料1トン当たり200g以下でなければならない。
ク フマル酸の飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、フマル酸として2.0%以下でなければならない。
ケ(ア) 鶏を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料は、飼料添加物であるβ-アポ-8'-カロチン酸エチルエステルを含んではならない。
(イ) 飼料添加物であるβ-アポ-8'-カロチン酸エチルエステルの飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、飼料1トン当たり80g以下でなければならない。
コ(ア) 鶏、さけ科魚類及び甲殻類を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料は、飼料添加物であるカンタキサンチンを含んではならない。
(イ) 飼料添加物であるカンタキサンチンの飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、鶏を対象とする飼料にあつては飼料1トン当たり8g以下、さけ科魚類及び甲殻類を対象とする飼料にあつては飼料1トン当たり80g以下でなければならない。
サ グルコン酸ナトリウムの飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、1.0%以下でなければならない。
シ 組換えDNA技術(組換えDNA(酵素等を用いて行うDNAの切断及び再結合の操作により作製されるDNAをいう。以下同じ。)を生細胞に移入し、これを増殖させる技術をいい、次に掲げるものを除く。以下同じ。)によつて得られた生物を含む飼料を製造する場合は、当該飼料は、その安全性につき、農林水産大臣の定めるところにより、農林水産大臣の確認を受けたものでなければならない。ただし、当該飼料が安全性の確保に支障がないものとして農林水産大臣が定める基準に適合する場合は、この限りでない。
(ア) 生細胞に移入された組換えDNAが当該生細胞と同一の分類学上の種に属する微生物のDNAのみからなるようにする技術
(イ) 組換えDNAが移入された生細胞の遺伝子の構成が自然界に存在する微生物の遺伝子の構成と同等となるようにする技術
ス 組換えDNA技術によつて得られた生物を利用して飼料を製造する場合は、当該飼料は、その安全性につき、農林水産大臣の定めるところにより、農林水産大臣の確認を受けたものでなければならない。
セ 次の表の第1欄に掲げる農薬(農薬取締法(昭和23年法律第82号)第2条第1項に規定する農薬をいう。以下同じ。)の成分である物質(その物質が化学的に変化して生成した物質を含む。以下同じ。)は、同表の第2欄に掲げる飼料の原料にそれぞれ同表の第3欄に定める量を超えて含まれてはならない。
| 第 1 欄 | 第 2 欄 | 第 3 欄 | ||||||||||
| γ-BHC | 牧草 | 0.4mg/kg | ||||||||||
| 2,4-ジクロロフェノキシ酢酸 |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
2mg/kg 2mg/kg 2mg/kg 0.05mg/kg 2mg/kg 2mg/kg 400mg/kg |
||||||||||
| BHC(α-BHC、β-BHC、γ-BHC及びδ-BHCの総和をいう。) | 牧草 | 0.02mg/kg |
||||||||||
| DDT(DDD及びDDEを含む。) | 牧草 | 0.1mg/kg | ||||||||||
| アセフェート | とうもろこし 牧草 |
0.5mg/kg 3mg/kg |
||||||||||
| アトラジン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.02mg/kg 0.02mg/kg 0.3mg/kg 0.2mg/kg 0.02mg/kg 0.02mg/kg 15mg/kg |
||||||||||
|
アラクロール |
えん麦 とうもろこし マイロ 牧草 |
0.1mg/kg 0.02mg/kg 0.05mg/kg 0.05mg/kg |
||||||||||
|
アルジカルブ |
えん麦 大麦 小麦 とうもろこし マイロ 牧草 |
0.2mg/kg 0.02mg/kg 0.02mg/kg 0.05mg/kg 0.2mg/kg 1mg/kg |
||||||||||
| アルドリン及びディルドリン(総和をいう。) | 牧草 | 0.02mg/kg | ||||||||||
| イソフェンホス | とうもろこし | 0.02mg/kg | ||||||||||
| イマザピック |
小麦 大豆 大豆油かす とうもろこし 牧草 |
0.05mg/kg 0.5mg/kg 0.5mg/kg 0.01mg/kg 3mg/kg |
||||||||||
| イマザピル |
大麦 小麦 大豆 大豆油かす とうもろこし 牧草 |
0.7mg/kg 0.05mg/kg 5mg/kg 7mg/kg 0.05mg/kg 30mg/kg |
||||||||||
| イミダクロプリド |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.04mg/kg 0.04mg/kg 0.2mg/kg 0.05mg/kg 0.04mg/kg 0.04mg/kg 0.5mg/kg |
||||||||||
| エチオン | 牧草 | 20mg/kg | ||||||||||
| エンドリン | 牧草 | 0.01mg/kg | ||||||||||
| カルタップ、チオシクラム及びベンスルタップ(総和をいう。) |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.7mg/kg |
||||||||||
| カルバリル |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
10mg/kg 5mg/kg 2mg/kg 0.1mg/kg 10mg/kg 5mg/kg 250mg/kg |
||||||||||
| カルベンダジム、チオファネート、チオファネートメチル及びベノミル(総和をいう。) |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.6mg/kg 0.6mg/kg 0.6mg/kg 0.7mg/kg 0.6mg/kg 0.6mg/kg 10mg/kg |
||||||||||
|
カルボフラン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.1mg/kg 0.2mg/kg 0.2mg/kg 0.05mg/kg 0.1mg/kg 0.1mg/kg 13mg/kg |
||||||||||
| キャプタン | とうもろこし | 10mg/kg | ||||||||||
| グリホサート |
えん麦 大麦 小麦 大豆 大豆油かす とうもろこし マイロ ライ麦 牧草 |
30mg/kg 30mg/kg 30mg/kg 20mg/kg 9mg/kg 5mg/kg 30mg/kg 30mg/kg 500mg/kg |
||||||||||
| グルホシネート |
大麦 小麦 とうもろこし |
0.5mg/kg 0.2mg/kg 0.1mg/kg |
||||||||||
| クロルピリホス |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.75mg/kg 0.2mg/kg 0.5mg/kg 0.1mg/kg 0.75mg/kg 0.01mg/kg 13mg/kg |
||||||||||
| クロルピリホスメチル |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 |
10mg/kg 6mg/kg 10mg/kg 7mg/kg 10mg/kg 7mg/kg |
||||||||||
| クロルフェンビンホス | 小麦 とうもろこし |
0.05mg/kg 0.05mg/kg |
||||||||||
| クロルプロファム |
えん麦 大麦 小麦 とうもろこし ライ麦 |
0.02mg/kg 0.02mg/kg 0.02mg/kg 0.05mg/kg 0.02mg/kg |
||||||||||
| ジカンバ |
えん麦 大麦 小麦 大豆 大豆油かす とうもろこし マイロ ライ麦 牧草 |
3mg/kg 7mg/kg 2mg/kg 10mg/kg 10mg/kg 0.5mg/kg 4mg/kg 0.1mg/kg 200mg/kg |
||||||||||
| ジクロルボス及びナレド(総和をいう。) |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 10mg/kg |
||||||||||
| ジクワット |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
2mg/kg 5mg/kg 2mg/kg 0.05mg/kg 2mg/kg 0.03mg/kg 100mg/kg |
||||||||||
| シハロトリン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.2mg/kg 0.2mg/kg 0.05mg/kg 0.04mg/kg 0.2mg/kg 0.02mg/kg 0.6mg/kg |
||||||||||
| シフルトリン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
2mg/kg 2mg/kg 2mg/kg (※令和9年1月7日より0.2mg/kg) 2mg/kg (※令和9年1月7日より0.05mg/kg) 3.5mg/kg 2mg/kg 50mg/kg |
||||||||||
| シマジン |
とうもろこし 牧草 |
0.3mg/kg 9mg/kg |
||||||||||
| ジメトエート |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.2mg/kg 0.04mg/kg 0.05mg/kg 1mg/kg 0.2mg/kg 0.2mg/kg 2mg/kg |
||||||||||
| ダイアジノン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.1mg/kg 0.1mg/kg 0.1mg/kg 0.02mg/kg 0.1mg/kg 0.1mg/kg 10mg/kg |
||||||||||
| チアベンダゾール |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.05mg/kg 0.05mg/kg 0.5mg/kg 0.05mg/kg 0.05mg/kg 0.05mg/kg 10mg/kg |
||||||||||
| デルタメトリン及びトラロメトリン(総和をいう。) |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
2mg/kg 2mg/kg 2mg/kg 2mg/kg 2mg/kg 2mg/kg 5mg/kg (※令和9年1月7日より1mg/kg) |
||||||||||
| テルブホス |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.05mg/kg 0.01mg/kg 0.01mg/kg 0.01mg/kg 0.05mg/kg 0.005mg/kg 1mg/kg |
||||||||||
| トリシクラゾール |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.02mg/kg 0.02mg/kg 0.02mg/kg 0.02mg/kg 0.02mg/kg 0.02mg/kg 5mg/kg |
||||||||||
| 二臭化エチレン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 |
0.01mg/kg 0.01mg/kg 0.1mg/kg 0.01mg/kg 0.01mg/kg 0.01mg/kg |
||||||||||
| パラコート |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.5mg/kg 0.05mg/kg 1.1mg/kg 0.1mg/kg 0.05mg/kg 0.05mg/kg 5mg/kg |
||||||||||
|
パラチオン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.08mg/kg 0.5mg/kg 0.3mg/kg 0.3mg/kg 0.08mg/kg 0.05mg/kg 5mg/kg |
||||||||||
| ピペロニルブトキシド |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 |
24mg/kg 24mg/kg 24mg/kg 24mg/kg 24mg/kg 24mg/kg |
||||||||||
| ピリミホスメチル |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 |
1mg/kg 1mg/kg 1mg/kg 1mg/kg 1mg/kg 1mg/kg |
||||||||||
| フィプロニル |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 |
0.002mg/kg 0.002mg/kg 0.002mg/kg 0.02mg/kg 0.01mg/kg 0.002mg/kg |
||||||||||
| フェニトロチオン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
6mg/kg 6mg/kg 15mg/kg 1mg/kg 6mg/kg 6mg/kg 10mg/kg |
||||||||||
| フェノブカルブ | 小麦 | 0.3mg/kg | ||||||||||
| フェントエート |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 |
0.4mg/kg 0.4mg/kg 0.4mg/kg 0.4mg/kg 0.4mg/kg 0.4mg/kg |
||||||||||
| フェンバレレート | 牧草 | 13mg/kg | ||||||||||
| フェンプロパトリン | 牧草 | 20mg/kg | ||||||||||
| ブロモキシニル |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.1mg/kg |
||||||||||
| ヘプタクロル | 牧草 | 0.02mg/kg | ||||||||||
| ペルメトリン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
2mg/kg 2mg/kg 2mg/kg 2mg/kg 2mg/kg 2mg/kg 55mg/kg |
||||||||||
| ベンタゾン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 3mg/kg |
||||||||||
| ペンディメタリン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草(アルファルファに限る。) 牧草(アルファルファを除く。) |
0.1mg/kg 0.2mg/kg 0.2mg/kg 0.2mg/kg 0.1mg/kg 0.2mg/kg 150mg/kg 2,000mg/kg |
||||||||||
| ホスメット |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.05mg/kg 0.05mg/kg 0.05mg/kg 0.05mg/kg 0.05mg/kg 0.05mg/kg 40mg/kg |
||||||||||
| ホレート |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.05mg/kg 0.05mg/kg 0.05mg/kg 0.05mg/kg 0.05mg/kg 0.05mg/kg 1.5mg/kg |
||||||||||
| マラチオン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
3mg/kg 2mg/kg 10mg/kg 2mg/kg 6mg/kg 2mg/kg 135mg/kg |
||||||||||
| メチダチオン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 牧草 |
0.2mg/kg 0.02mg/kg 0.02mg/kg 0.1mg/kg 0.2mg/kg 0.02mg/kg 12mg/kg |
||||||||||
| メトプレン |
えん麦 大麦 小麦 とうもろこし マイロ ライ麦 |
5mg/kg 5mg/kg 5mg/kg 5mg/kg 5mg/kg 5mg/kg |
||||||||||
|
備考
1 第2欄における次に掲げる飼料の原料は、それぞれ次に定める部位をいう。
2 「牧草」には、乾燥して水分含量を低下させたもの及びサイレージ(牧草(乾燥して水分含量を低下させたものを含む。)をサイロ又は適当な容器に詰め、乳酸発酵させて調製する飼料をいう。)を含む。
3 第2欄に掲げる飼料の原料が牧草である場合において、第1欄に掲げる農薬の成分である物質の当該飼料の原料中の含有量を算出するに当たつては、当該飼料の原料中の水分の含有量が10%を超えるときは、その超える量を当該飼料の原料の量から除外するものとする。
|
||||||||||||
ソ 次の表の第1欄に掲げる農薬の成分である物質は、同表の第2欄に掲げる家畜等(法第2条第1項に規定する家畜等をいう。以下同じ。)を対象とする飼料にそれぞれ同表の第3欄に定める量を超えて含まれてはならない。
| 第 1 欄 | 第 2 欄 | 第 3 欄 |
| γーBHC | 牛、馬、めん羊、山羊及び鹿 豚 鶏及びうずら |
0.4mg/kg 0.05mg/kg 0.05mg/kg |
| BHC(α-BHC、β-BHC、γ-BHC及びδ-BHCの総和をいう。) | 牛、馬、めん羊、山羊及び鹿 豚 鶏及びうずら |
0.005mg/kg 0.005mg/kg 0.005mg/kg |
| DDT(DDD及びDDEを含む。) | 牛、馬、めん羊、山羊及び鹿 豚 鶏及びうずら |
0.1mg/kg 0.1mg/kg 0.1mg/kg |
| アルドリン及びディルドリン(総和をいう。) | 牛、馬、めん羊、山羊及び鹿 豚 鶏及びうずら |
0.02mg/kg 0.02mg/kg 0.02mg/kg |
| エンドリン | 牛、馬、めん羊、山羊及び鹿 豚 鶏及びうずら |
0.01mg/kg 0.01mg/kg 0.01mg/kg |
| フェンバレレート | 牛、めん羊、山羊及び鹿 豚 鶏及びうずら |
8mg/kg 4mg/kg 0.5mg/kg |
| ヘプタクロル | 牛、馬、めん羊、山羊及び鹿 豚 鶏及びうずら |
0.02mg/kg 0.02mg/kg 0.02mg/kg |
タ ギ酸カルシウムの飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、ギ酸カルシウムとして1.5%以下でなければならない。
チ 二ギ酸カリウムの飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、二ギ酸カリウムとして1.8%以下でなければならない。
ツ 25-ヒドロキシコレカルシフェロールの飼料(飼料を製造するための原料又は材料を除く。以下ツにおいて同じ。)中の含有量は、牛を対象とする飼料にあつては飼料1トン当たり100mg以下、豚を対象とする飼料にあつては飼料1トン当たり50mg以下、鶏を対象とする飼料にあつては飼料1トン当たり80mg以下でなければならない。
テ グアニジノ酢酸の飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、グアニジノ酢酸として0.06%以下でなければならない。
ト 安息香酸の飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、安息香酸として0.5%以下でなければならない。
ナ 3-ニトロオキシプロパノールの飼料(飼料を製造するための原料又は材料を除く。)中の含有量は、3-ニトロオキシプ ロパノールとして0.015%以下でなければならない。
ニ(ア) 牛を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料は、飼料添加物であるカシューナッツ殻液を含んではならない。
(イ) 牛を対象とする飼料(飼料を製造するための原料又は材料を除く。)中のカシューナッツ殻液の含有量は、カシューナッツ殻液として0.1%以下でなければならない。
ヌ アセチルシステインの飼料(飼料製造するための原料又は材料を除く。)中の含有量は、アセチルシステインとして0.0014%以下でなければならない。
(2) 飼料一般の製造の方法の基準
ア 有害な物質を含み、若しくは病原微生物により汚染され、又はこれらの疑いがある原料又は材料を用いてはならない。
イ 成分について規格が定められた飼料又は飼料添加物を原料又は材料とする場合においては、当該規格に合うもの(法第5条第1項の検定を要するものにあつては、当該検定に合格したものに限る。)を用いなければならない。
ウ 次の表の同一欄内の2以上の飼料添加物は、同一飼料に用いてはならない。
| 第1欄 | アンプロリウム・エトパベート、アンプロリウム・エトパベート・スルファキノキサリン、サリノマイシンナトリウム、センデュラマイシンナトリウム、ナイカルバジン、ナラシン、モネンシンナトリウム、ラサロシドナトリウム |
| 第2欄 | クエン酸モランテル |
| 第3欄 | アビラマイシン、エンラマイシン、ノシヘプタイド、フラボフォスフォリポール |
エ ギ酸は、牛、馬、豚、鶏及びうずら対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
オ プロピレングリコールは、体重がおおむね30kg以内の豚を対象とする飼料及び生後おおむね3月以内の牛を対象とする飼料以外の飼料には用いてはならない。
カ フマル酸は、体重がおおむね70kg以内の豚(種豚育成中のものを除く。)を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
キ 次の表の左欄に掲げる飼料添加物は、同表の右欄に掲げる対象飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
| 飼料添加物名 | 対象飼料 |
|---|---|
| エンテロコッカス フェカーリス(クロストリジウム ブチリカム(その2)製剤及びバチルス サブチルス(その4)製剤と混合して使用する場合に限る。) | 牛用、豚用、鶏用及びうずら用 |
| エンテロコッカス フェシウム(その1)(ラクトバチルス アシドフィルス(その1)製剤と混合して使用する場合に限る。) | 牛用、鶏用及びうずら用 |
| エンテロコッカス フェシウム(その2)(ラクトバチルス アシドフィルス(その6)製剤と混合して使用する場合に限る。) | 豚用 |
| エンテロコッカス フェシウム(その3) | 牛用、豚用、鶏用及びうずら用 |
| エンテロコッカス フェシウム(その4)(ビフィドバクテリウム サーモフィラム(その2)製剤及びラクトバチルス アシドフィルス(その5)製剤と混合して使用する場合に限る。) | 牛用及び豚用 |
| クロストリジウム ブチリカム(その1) | 牛用、馬用、豚用、鶏用及びうずら用 |
| バチルス コアグランス | 豚用 |
| バチルス サブチルス(その1) | 牛用、馬用、豚用、鶏用及びうずら用 |
| バチルス サブチルス(その2) | 牛用、馬用、豚用、鶏用及びうずら用 |
| バチルス サブチルス(その3) | 牛用、馬用、豚用、鶏用及びうずら用 |
| バチルス サブチルス(その5) | 豚用及び鶏用 |
| バチルス セレウス | 牛用、豚用、鶏用、うずら用及び養殖水産動物(飼料の安全性の確保及び品質の改善に関する法律施行令第1条第4号に掲げる動物をいう。以下同じ。)用 |
| バチルス バディウス | 豚用 |
| ビフィドバクテリウム サーモフィラム(その1)(ラクトバチルス サリバリウス製剤と混合して使用する場合に限る。) | 鶏用及びうずら用 |
| ビフィドバクテリウム サーモフィラム(その3) | 牛用及び豚用 |
| ビフィドバクテリウム サーモフィラム(その4) | 牛用 |
| ビフィドバクテリウム シュードロンガム(その1) | 豚用 |
| ビフィドバクテリウム シュードロンガム(その2) | 牛用及び豚用 |
| ラクトバチルス アシドフィルス(その2) | 鶏用及びうずら用 |
| ラクトバチルス アシドフィルス(その3) | 牛用及び馬用 |
| ラクトバチルス アシドフィルス(その4) | 豚用 |
| ラクトバチルス アシドフィルス(その5) | 牛用、馬用及び豚用 |
| ラクトバチルス アシドフィルス(その6) | 豚用 |
ク(ア) 製造に2以上の原料又は材料を用いる場合には、これらを原料又は材料として製造される飼料が均質なものとなるようにしなければならない。
(イ) 飼料添加物を用いる場合には、当該飼料添加物の効果が阻害されないような製造方法によらなければならない。
ケ グルコン酸ナトリウムは、体重がおおむね70kg以内の豚(種豚育成中のものを除く。)を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
コ 組換えDNA技術によつて得られた微生物を利用して飼料を製造する場合は、農林水産大臣が定める基準に適合する旨の農林水産大臣の確認を得た方法で製造しなければならない。
サ グルコン酸カルシウムは、牛、めん羊、山羊及び鹿(以下「牛等」という。)並びに馬を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
シ ギ酸カルシウムは、体重がおおむね70kg以内の豚(種豚育成中のものを除く。)を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ス 二ギ酸カリウムは、体重がおおむね70kg以内の豚(種豚育成中のものを除く。)を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
セ 25-ヒドロキシコレカルシフェロールは、牛、豚及び鶏を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ソ フィターゼ(その2の(3))は、豚及び鶏を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
タ L-カルニチンは、種豚(体重がおおむね120kgを超えたものに限る。)を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
チ アルカリ性プロテアーゼ(その3)は、豚及び鶏を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ツ グアニジノ酢酸は、ブロイラーを対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
テ フィターゼ(その2の(4))は、豚、鶏及びうずらを対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ト フィターゼ(その2の(5))は、豚、鶏及びうずらを対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ナ フィターゼ(その2の(6))は、豚、鶏、うずら、魚類及び甲殻類を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ニ ムラミダーゼは、豚及び鶏を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ヌ 安息香酸は、豚を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ネ フィターゼ(その2の(7))は、豚、鶏及びうずらを対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ノ 3-ニトロオキシプロパノールは、牛を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ハ 2―デアミノ―2―ヒドロキシメチオニンイソプロピルエステルは、牛を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ヒ アミラーゼ(その3)は、牛、豚及び鶏を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
フ フィターゼ(その2の(8))は、豚、鶏、うずら、魚類及び甲殻類を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
ヘ アセチルシステインは、鶏(ブロイラーを除く。)を対象とする飼料(飼料を製造するための原料又は材料を含む。)以外の飼料に用いてはならない。
(3) 飼料一般の使用の方法の基準
ア 有害な物質を含み、若しくは病原微生物により汚染され、又はこれらの疑いがある飼料は、使用してはならない。
イ(ア) 表示の基準に基づき対象家畜等(当該飼料を使用することができる家畜等をいう。以下同じ。)が表示されている飼料は、当該対象家畜等以外の家畜等に対し使用してはならない。
(イ) (1)のウの表に掲げる飼料添加物を含む同表の対象飼料は、搾乳中の牛又は産卵中の鶏若しくはうずら並びに食用を目的として屠殺する前7日間の牛(生後おおむね6月を超えた肥育牛を除く。)、豚、鶏又はうずらに使用してはならない。
(ウ) 綿実油かすを原料とする飼料は、養殖水産動物に対し使用してはならない。
ウ 表示の基準に基づき使用上の注意事項が表示されている飼料は、当該使用上の注意事項を遵守して使用しなければならない。
エ (2)のウの表の同一欄内の2以上の飼料添加物を含む飼料は、使用してはならない。
オ (2)のウの表の飼料添加物を含む飼料は、当該飼料添加物が掲げられている欄内の他の飼料添加物を含む飼料と併用してはならない。
カ 飼料は、使用後に次に掲げる事項を帳簿に記載して保存するよう努めなければならない。
(ア) 当該飼料を使用した年月日
(イ) 当該飼料を使用した場所
(ウ) 当該飼料を使用した家畜等の種類
(エ) 当該飼料の名称
(オ) 当該飼料の使用量
(カ) 当該飼料を譲り受けた年月日及び相手方の氏名又は名称
(4) 飼料一般の保存の方法の基準
ア 有害な物質を含み、若しくは病原微生物により汚染され、又はこれらの疑いがある場所に保存し、又は有害な物質を含み、若しくは病原微生物により汚染され、又はこれらの疑いがある容器若しくは包装材料を用いて保存してはならない。
イ 表示の基準に基づき保存上の注意事項が表示されている飼料は、当該保存上の注意事項を遵守して保存しなければならない。
(5) 飼料一般の表示の基準
ア 輸出用又は試験研究用の飼料には、「輸出用」又は「試験研究用」という文字を表示しなければならない。
イ 飼料(飼料添加物を含むものに限る。)には、次に掲げる事項を表示しなければならない。
(ア) 飼料の名称
(イ) 製造(輸入)年月
(ウ) 製造(輸入)業者の氏名又は名称及び住所
(エ) 製造事業場の名称及び所在地(輸入に係るものにあっては、輸入先国名)
(オ) (1)のウに掲げる表、(1)のキの(ア)、ケの(ア)、コの(ア)、ニの(ア)及びヌ、(2)のエからカまで、(2)のキに掲げる表並びに(2)のケ及びサからヘまでに対象とする家畜等が定められている飼料にあっては、対象家畜等
(カ) 飼料添加物を含む飼料にあっては含有する飼料添加物の名称及び量
(キ) (3)のイの(イ)に規定する飼料にあっては、(3)のイの(イ)に規定する趣旨
(ク) サリノマイシンナトリウム、モネンシンナトリウム又はラサロシドナトリウムを含む牛用の肥育期用飼料にあっては、次の文字
使用上の注意
1 生後おおむね6月を超えた肥育牛(搾乳中のものを除く。)以外には使用しないこと(特に馬に給与すると障害を起こしやすいので注意すること。)。
2 新たにこの飼料の給与を開始しようとする場合は、給与量を段階的に増加させていくこと。
(ケ) サリノマイシンナトリウム又はモネンシンナトリウムを含む牛用の幼令期用飼料にあっては、次の文字
使用上の注意
1 生後おおむね3月を超え6月以内の幼令牛以外には使用しないこと(特に馬に給与すると障害を起こしやすいので注意すること。)。
2 新たにこの飼料の給与を開始しようとする場合は、給与量を段階的に増加させていくこと。
(コ) モネンシンナトリウムを含む牛用のほ乳期用飼料にあつては、次の文字
使用上の注意
1 生後おおむね3月以内の牛以外には使用しないこと(特に馬に給与すると障害を起こしやすいので注意すること。)。
2 新たにこの飼料の給与を開始しようとする場合は、給与量を段階的に増加させていくこと。
(サ) ナイカルバジンを含むブロイラー用の前期用飼料にあっては、次の文字
使用上の注意
ふ化後おおむね8週間以内に出荷するブロイラーに使用する場合は、この飼料を給与した場所と異なる場所で、当該ブロイラーを食用を目的としてと殺する前7日間以上飼養すること。
(注)1 飼料添加物の名称の表示については、法第2条第3項の規定に基づき農林水産大臣が飼料添加物を指定する場合に、当該飼料添加物の名称として用いるものによるものとする。ただし、広く一般に使用されている名称を有する飼料添加物にあつては、その名称をもつてこれに代えることができる。
2 飼料添加物の量の表示については、次による。
1) (1)のウの表に掲げる飼料添加物については、同表に掲げる単位を用いて表示するものとする。
2) プロピオン酸、プロピオン酸カルシウム及びプロピオン酸ナトリウム(飼料を製造するための原料又は材料に含有されている場合に限る。)については、プロピオン酸としての含有率を、ギ酸(飼料を製造するための原料又は材料に含有されている場合に限る。)については、ギ酸としての含有率を、フマル酸(飼料を製造するための原料又は材料に含有されている場合に限る。)については、フマル酸としての含有率をそれぞれパーセントで表示するものとする。
3) エトキシキン、ジブチルヒドロキシトルエン及びブチルヒドロキシアニソール(飼料を製造するための原料又は材料に含有されている場合に限る。)については、それぞれの有効成分の合計の含有率をパーセントで表示するものとする。
4) 飼料添加物としてのアスタキサンチン(飼料を製造するための原料又は材料に含有されている場合に限る。)については、魚類を対象とする飼料にあつては飼料1トン当たり100g、甲殻類を対象とする飼料にあつては飼料1トン当たり200gを超えて含有されている場合に限り、含有率をパーセントで表示するものとする。
5) 飼料添加物としてのβ-アポ-8'-カロチン酸エチルエステル(飼料を製造するための原料又は材料に含有されている場合に限る。)については、飼料1トン当たり80gを超えて含有されている場合に限り、含有率をパーセントで表示するものとする。
6) 飼料添加物としてのカンタキサンチン(飼料を製造するための原料又は材料に含有されている場合に限る。)については、鶏を対象とする飼料にあつては飼料1トン当たり8g、さけ科魚類及び甲殻類を対象とする飼料にあつては飼料1トン当たり80gを超えて含有されている場合に限り、含有率をパーセントで表示するものとする。
7) その他の飼料添加物については、量の表示を要しない。
3 飼料又は飼料添加物の製造業者のみに販売する場合には、農林水産大臣の承認を受けて「製造業者専用」の文字を表示し、上記の表示すべき事項の一部を省略することができる。
ウ 表示は、法第32条第1項の規定に基づく表示の基準に従い行う表示に準じて行うものとする。
(1) 動物由来たん白質又は動物由来たん白質を原料とする飼料の成分規格
家畜等を対象とする飼料は、動物由来たん白質(ほ乳動物由来たん白質(ほ乳動物に由来するたん白質をいい、乳及び乳製品を除く。以下同じ。)、家きん由来たん白質(家きんに由来するたん白質をいい、卵及び卵製品を除く。以下同じ。)又は魚介類由来たん白質(魚介類に由来するたん白質をいう 。以下同じ。)をいう。以下同じ。)を含んではならない。ただし、次の表の第1欄に掲げる家畜等を対象とする飼料は、それぞれ同表の第2欄に掲げる動物由来たん白質を含むことができる。
| 第1欄 | 第2欄 |
| 牛等 |
ア 次の(ア)から(オ)までのいずれかに該当するゼラチン又はコラーゲンであつて、これら以外のたん白質の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(以下「確認済ゼラチン等」という。)
(ア) ほ乳動物(反すう動物にあつては、牛、めん羊及び山羊に限る。)の皮に由来するものであること。
(イ) ほ乳動物(反すう動物を除く。)の骨に由来するものであつて、次の工程の全てを経て処理されたもの又はこれと同等以上の処理がされたものであること。
a 脱脂
b 酸による脱灰
c 酸処理又はアルカリ処理
d ろ過
e 138℃以上で4秒間以上の殺菌処理
(ウ) 牛の骨(頭蓋骨及び脊柱(背根神経節を含み、胸椎横突起、腰椎横突起、仙骨翼及び尾椎を除く。)を除く。)に由来するものであつて、(イ)のaからeまでに掲げる工程の全てを経て処理されたもの又はこれと同等以上の処理がされたものであること。
(エ) めん羊又は山羊の骨(頭蓋骨及び脊柱を除く。)に由来するものであつて、(イ)のaからeまでに掲げる工程の全てを経て処理されたもの又はこれと同等以上の処理がされたものであること。
(オ) 家きん又は魚介類に由来するものであること。
|
| 馬、豚、鶏、うずら又は養殖水産動物 |
ア 確認済ゼラチン等
イ 豚(いのししを含む。以下この表において同じ。)又は馬に由来する血粉又は血しようたん白質であつて、これら以外のたん白質の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(以下「確認済豚血粉等」という。)
ウ 豚に由来する肉骨粉、加水分解たん白質又は蒸製骨粉であつて、これら以外のたん白質の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(以下「確認済豚肉骨粉等」という。)
エ 馬に由来する肉骨粉、加水分解たん白質又は蒸製骨粉であつて、これら以外のたん白質の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(以下「確認済馬肉骨粉等」という。)
オ 豚、馬又は家きんに由来する原料を混合して製造された肉骨粉、加水分解たん白質、蒸製骨粉、血粉又は血しようたん白質であつて、豚、馬又は家きん以外の動物に由来するたん白質の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(以下「確認済原料混合肉骨粉等」という。)
カ 家きん由来たん白質のうち、チキンミール、フェザーミール、血粉又は血しようたん白質であつて、これら以外のたん白質の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(以下「確認済チキンミール等」という。)
キ 家きん由来たん白質のうち、加水分解たん白質又は蒸製骨粉であつて、これら以外のたん白質の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(以下「確認済家きん加水分解たん白質等」という。)
ク 魚介類由来たん白質であつて、ほ乳動物由来たん白質及び家きん由来たん白質(確認済ゼラチン等を除く。)の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(以下「確認済魚介類由来たん白質」という。)
ケ 牛、豚、めん羊、山羊、馬又は家きんに由来する血粉又は血しようたん白質(月齢が30月を超える牛(出生の年月日から起算して30月を経過した日の翌日以後のものをいう。)の脊柱(背根神経節を含み、頚椎横突起、胸椎横突起、腰椎横突起、頚椎棘突起、胸椎棘突起、腰椎棘突起、仙骨翼、正中仙骨稜及び尾椎を除く。以下同じ。)及びと畜場法(昭和28年法律第114号)第14条の検査を経ていない牛の部位(以下「牛の脊柱等」という。)並びに当該検査を経ていないめん羊又は山羊の部位及びと畜場法施行規則(昭和28年厚生省令第44号)別表第一のめん羊又は山羊の部位(以下「めん山羊の部位」という。)が混入していないものに限る。)であつて、これら以外のたん白質の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(イ、オ及びカに掲げるものを除く。以下「確認済牛血粉等」という。)
コ 牛、豚、めん羊、山羊、馬又は家きんに由来する肉骨粉、加水分解たん白質又は蒸製骨粉(牛の脊柱等及びめん山羊の部位が混入していないものに限る。)であつて、これら以外のたん白質の製造工程と完全に分離された工程において製造されたことについて農林水産大臣の確認を受けたもの(ウからキまでに掲げるものを除く。以下「確認済牛肉骨粉等」という。
サ 食品循環資源(食品循環資源の再生利用等の促進に関する法律(平成12年法律第116号)第2条第3項に規定する食品循環資源をいう。以下同じ。)に含まれる動物由来たん白質であつて、農林水産大臣が指定するもの
|
| 蜜蜂 |
ア 確認済ゼラチン等
イ 確認済豚血粉等
ウ 確認済チキンミール等
エ 確認済魚介類由来たん白質
|
(2) 動物由来たん白質又は動物由来たん白質を原料とする飼料の製造の方法の基準
ア 動物由来たん白質は、(1)の表の第1欄に掲げる家畜等を対象として、それぞれ同表の第2欄に掲げる動物由来たん白質が含まれる飼料を用いる場合を除き、家畜等を対象とする飼料(飼料を製造するための原料又は材料を含む。)に用いてはならない。
イ 牛等を対象とする飼料(飼料を製造するための原料又は材料を含む。)は、動物由来たん白質(確認済ゼラチン等を除く。)を含む飼料(飼料を製造するための原料又は材料を含む。)の製造工程と完全に分離された工程において製造されなければならない。
ウ 確認済牛血粉等又は確認済牛肉骨粉等を含む馬、豚、鶏、うずら又は養殖水産動物を対象とする飼料は、牛等を対象とする飼料(飼料を製造するための原料又は材料を含む。)の製造工程と完全に分離していることについて農林水産大臣の確認を受けた工程において製造されなければならない。
(3) 動物由来たん白質又は動物由来たん白質を原料とする飼料の使用の方法の基準
動物由来たん白質を含む飼料は、(1)の表の第1欄に掲げる家畜等を対象として、それぞれ同表の第2欄に掲げる動物由来たん白質が含まれる飼料を使用する場合を除き、家畜等に対し使用してはならない。
(4) 動物由来たん白質又は動物由来たん白質を原料とする飼料の保存の方法の基準
動物由来たん白質を含む飼料は、(1)の表の第1欄に掲げる家畜等を対象として、それぞれ同表の第2欄に掲げる動物由来たん白質が含まれる飼料を保存する場合を除き、家畜等を対象とする飼料(飼料を製造するための原料又は材料を含む。)に混入しないように保存しなければならない。
(5) 動物由来たん白質又は動物由来たん白質を原料とする飼料の表示の基準
ア 確認済豚血粉等、確認済豚肉骨粉等、確認済馬肉骨粉等、確認済チキンミール等、確認済家きん加水分解たん白質等、確認済魚介類由来たん白質、確認済原料混合肉骨粉等、確認済牛血粉等若しくは確認済牛肉骨粉等又はこれらを原料とする飼料には、次に掲げる事項を表示しなければならない。
(ア) 飼料の名称
(イ) 製造(輸入)年月
(ウ) 製造(輸入)業者の氏名又は名称及び住所
(エ) 製造事業場の名称及び所在地(輸入に係るものにあつては、輸入先国名)
イ 確認済豚血粉等、確認済豚肉骨粉等、確認済馬肉骨粉等、確認済チキンミール等、確認済家きん加水分解たん白質等、確認済魚介類由来たん白質、確認済原料混合肉骨粉等、確認済牛血粉等若しくは確認済牛肉骨粉等又はこれらを原料とする飼料には、次の文字を表示しなければならない。
使用上及び保存上の注意
1 この飼料は、牛、めん羊、山羊及び鹿には使用しないこと(牛、めん羊、山羊又は鹿に使用した場合は処罰の対象となるので注意すること。)。
2 この飼料は、牛、めん羊、山羊及び鹿を対象とする飼料(飼料を製造するための原料又は材料を含む。)に混入しないよう保存すること。
3 落花生油かす又は落花生油かすを原料とする飼料の成分規格及び使用の方法等の基準
(1) 落花生油かす又は落花生油かすを原料とする飼料の成分規格
ア 落花生油かすのアフラトキシンB1の含有量は1mg/kgを超えてはならない。この場合のアフラトキシンB1の定量法は、次に掲げる定量法A又は定量法Bによるものとする。
定量法A
a 分析機器
1) 振とう機(分液漏斗用)
2) 薄層クロマトグラフ装置
3) けい光検出装置 紫外線(365nm)を発生し、受光面における紫外線強度が1,000μw/cm2であるもの。
b 試薬及び試薬の調製
1) n-ヘキサン、アセトン及びクロロホルム 試薬特級で波長365nm付近の紫外線によってけい光を発生しないもの。
2) 無水硫酸ナトリウム 120℃で2時間乾燥したもの。
3) 薄層クロマトグラフィー用吸着剤 ワコーゲルB-O又はこれと同等の分離能を有するもの。
4) 標準アフラトキシン液
ア 標準アフラトキシン混合液 アフラトキシンB1、B2、G1及びG2標準品各5.0mgをおのおのベンゼン-アセトニトリル(98+2)500mLに溶かし、標準原液とする。使用時に、各標準原液の一定量を混合し、クロロホルムで希釈し、1mL当たりアフラトキシンB1、B2、G1及びG2各0.2μgの標準アフラトキシン混合液を調製する。
イ 標準アフラトキシンB1液 使用時に、アの標準アフラトキシンB1原液の一定量をとりクロロホルムで希釈し、1mL当たり0.2μgアフラトキシンB1の標準アフラトキシンB1液を調製する。
5) 展開液 クロロホルム-アセトン-n-ヘキサン(100+5+5)
c 薄層板の調製
薄層クロマトグラフィー用ワコーゲルB-O 30gに水60mLを加え十分に混合して、アプリケーターで薄層クロマト用ガラス板(20×20cm)に0.25mmの厚さに塗布し、風乾した後、110~120゚で2時間加熱して活性化する。
d 操作
1) 試料液の調製
試料20g(1000μmの網ふるいを通過させたもの)を500mLの分液漏斗にとり、水10mL及びクロロホルム100mLを加えて振とう機で30分間振り混ぜる。クロロホルム層をろ紙でろ過して試料液とする。
2) 展開分離
薄層板の一辺から3cm離れた位置をベースラインとし、ベースライン上に試料液を5、10、15、20μL及び標準アフラトキシン混合液及び標準アフラトキシンB1液各10μLマイクロシリンジで1~1.5cm間隔にスポットし、展開液
の先端が10cm以上になるよう展開する。
展開した後薄層板をとり出し、風乾して展開液を揮散させる。
e アフラトキシンB1量の測定
展開し風乾した薄層板をけい光検出装置の紫外灯の直下に置き、試料液の展開スポットに標準アフラトキシンB1のRfと一致するけい光スポットがあるかを判定する。
すべての試料液の展開スポットにアフラトキシンB1によるけい光が認められた場合には、試料液の数mLをクロロホルムで適宜希釈して、その液の10又は15μLの展開スポットに確認し得る最低のけい光強度(検出限界量という。以下同じ。)が得られるよう一定容量に調製する。
この分析条件におけるアフラトキシンB1の検出限界量は、0.4×10-9gである。
f アフラトキシンB1量の算出
アフラトキシンB1量は、次式により算出する。
アフラトキシンB1(μg/kg)=(400×S)÷(W×V)
S 試料液量(mL) 試料液を希釈した場合は、希釈率によって換算すること。
W 落花生油かすの量(20g)
V 検出限界量を示した試料液のスポット量(μL)
注
1) あらかじめ、使用する吸着剤を用いて定量法A・cに従つて薄層板を調製し、これに標準アフラトキシン混合液及び標準アフラトキシンB1液各10μLをスポットして定量法A・d・2)により展開し、展開スポットの分離の良否を確認する。アフラトキシンB1の展開スポットが他の展開スポットと完全に分離する吸着剤及びこれと同一製造ロットの吸着剤を定量操作に用いる。
2) 各標準液は、適当なかつ色共せんフラスコに入れ、mg単位まで目方を測つた後、0゚以下で保存する。使用時に、目方の減少がなく従つて濃度変化のないことを確認する。
3) 薄層クロマト用ガラス板に付着した油脂類は、吸着剤の塗布に支障があるので、エーテル・メタノール混合液を含ませた脱脂綿でふきとる。
4) 薄層板は、使用前に吸着剤の厚さの均一性、ひび割れの有無等を調べ、不良なものは使用しない。
5) ろ液に水が含まれる場合は無水硫酸ナトリウム約10gで脱水する。
6) 展開槽は、ふたのあるガラス又はステンレス製(縦25cm、横15cm、深さ30cm程度のもの)のものを用い、その内部に収める展開液用小容器(縦22cm、横5cm、深さ5cm)を用意する。展開に当たつては、展開液を深さ2cmまで展開用小容器に加え、これを展開槽に収めた後直ちに薄層板を挿入し、展開を行う。
7) スポットの直径は、5mm以下になるようにする。
8) 展開温度は、22~25゚とする。
9) 展開は、遮光して行う。
10) 周縁効果の影響を受ける場合は、標準液及び試料液の位置を変えて、繰り返し行う。
11) 展開スポットのテーリング又は拡散がはなはだしい場合は、別に調製した薄層板で分析を繰り返す。
12) 試料の展開スポットにけい光が認められない場合は、この試料のアフラトキシンB1量は100μg/kg以下である。100μg/kg以下のものについて測定する場合は、試料液の一定量を減圧(50゚以下)で濃縮又は乾固して、これにクロロホルムを加え、溶かして一定容量とし、これを定量法A・d・2)に従つてスポットし、その試料液10又は15μLの展開スポットに検出限界量のけい光が得られるように調製する。
13) けい光の有無の判定は、必ず複数の測定者によって行う。
14) 分析条件が異なればアフラトキシンB1の検出限界量は当然異なるので、ここに指定する分析条件と異なる条件で分析を行う場合は、標準アフラトキシンB1液を用いて検出限界量を別に定めなければならない。
付記 アフラトキシンは強力な発がん物質であることから、特に標準アフラトキシンB1液の調製の際は、その取扱いを慎重に行うこと。必要に応じ防塵マスク、ゴム手袋を装着すること。また、実験台、使用器具等は、使用後0.5~1%の次亜塩素酸ナトリウム液で洗浄又は拭清する。手先にアフラトキシンが付着したときは、上記次亜塩素酸ナトリウム液と石けんで十分に洗浄する。
定量法B
a 分析機器
1) 分光けい光デンシトメーター
2) その他の分析機器は、定量法A・aによる。
b 試薬及び試薬の調製
1) 試薬及び展開液は、定量法A・bによる。
2) 標準アフラトキシン液
ア 標準アフラトキシン混合液 定量法A・b・4)・イによる。
イ 標準アフラトキシンB1液 アフラトキシンB1標準品5.0mgをベンゼン-アセトニトリル(98+2)500mLに溶かし、標準原液とする。使用時に、標準原液の一定量をとりクロロホルムで希釈し、1mL当たり0.1、0.4、0.6及び1.0μgのアフラトキシンB1濃度の標準アフラトキシンB1液を調製する。
c 操作
定量法A・d・1)で調製した試料液の一定量(10~20μL)及び標準アフラトキシン混合液10μLを薄層板にスポットし、同時に検量線作成のため、標準アフラトキシンB1液を4濃度段階にスポットする。
薄層板の調製及び展開分離の方法は、定量法A・c及びA・d・2)による。
d 定量
展開し風乾した後、デンシトメーター(カットフイルター390nm、励起波長365nm、けい光波長430nm)により標準アフラトキシンB1液の展開スポット及び試料液のアフラトキシンB1スポットのけい光強度を測定し、標準アフラトキシンB1液の展開スポットのけい光強度から作成した検量線によって試料液中のアフラトキシンB1量を求め、試料中のアフラトキシンB1量を算出する。
イ 落花生油かすを原料とすることができる飼料の種類及びその配合割合は、次の表のとおりとする。
| 飼料の種類 | 配合割合 |
|---|---|
| 鶏用(幼すう用及びブロイラー前期用を除く。)飼料 | 4%以下 |
| 豚用(ほ乳期用を除く。)飼料 | 4%以下 |
| 搾乳牛用飼料 | 2%以下 |
| 牛用(ほ乳期用及び搾乳牛用を除く。)飼料 | 4%以下 |
注 搾乳牛とは、生後おおむね18月を超える搾乳の用に供する牛をいう。4の(1)のイにおいて同じ。
(2) 落花生油かすの使用の方法の基準
落花生油かすのみを単体で使用してはならない。
(3) 落花生油かす又は落花生油かすを原料とする飼料の表示の基準
ア 落花生油かす又は落花生油かすを原料とする飼料には、次に掲げる事項を表示しなければならない。
(ア) 飼料の名称
(イ) 製造(輸入)年月
(ウ) 製造(輸入)業者の氏名又は名称及び住所
(エ) 製造事業場の名称及び所在地(輸入に係るものにあつては、輸入先国名)
イ 落花生油かすを原料とする飼料には、次に掲げる事項を表示しなければならない。
(ア) 対象家畜等
(イ) 落花生油かすの配合割合
4 尿素若しくはジウレイドイソブタン又はこれらを原料とする飼料の成分規格及び製造の方法等の基準
(1) 尿素及びジウレイドイソブタン並びにこれらを原料とする飼料の成分規格
ア 尿素及びジウレイドイソブタンの成分規格は、次の表のとおりとする。
| 種類 | 尿素 | ジウレイドイソブタン |
|---|---|---|
| 事項 | ||
| 純度 水分 ビウレット 尿素 重金属 |
97%以上 0.5%以下 1.0%以下 - 10mg/kg以下 |
93%以上 2.0%以下 - 3.0%以下 10mg/kg以下 |
この場合の尿素及びジウレイドイソブタンの試験法は、それぞれ次のとおりとする。
○尿素の試験法
A 純度
ケルダール法によって求めた窒素の量からビウレット性窒素の量を減じ、純度を算出する。
尿素の純度(%)={(a-b)÷46.65}×100
a ケルダール法によって求めた窒素の量
b ビウレット性窒素の量
46.65 尿素中の窒素量の理論値
B 水分
試料約5gをひよう量ざらに正確にとり、75±1゚で4時間乾燥し、その減量を水分とする。
C ビウレット
a 試薬の調製
1) 標準ビウレット性窒素液
ビウレット〔(CO・NH2)2NH〕(110゚で恒量になるまで乾燥したもの)0.9813gを100mLのメスフラスコにとり、水に溶かし更に標線まで水を加える(この液1mLは、ビウレット性Nとして4mgを含有する)。
2) 硫酸銅液
硫酸銅15gを水に溶かして1Lとし、必要があればろ過する。
b 試料液の調製
試料約1~10g(ビウレット性Nとして20~60mgがよい。)を100mLのメスフラスコに正確にとり、水約50mLを加えて溶かす。
c 定量
試料液に4%水酸化ナトリウム液20mLを加え、更に硫酸銅液20mLを加えて発色させ、標線まで水を加えてよく振り混ぜ、約30分間放置後、遠心機により沈でんを分離し、その上澄み液をとり波長540nm付近の吸光度を測定する。別に標準ビウレット性窒素液の各種一定量について、試料液の場合と同一条件で操作して作成した検量線からビウレット性窒素〔N〕の量を求める。これに係数2.4531を乗じてビウレットの量とする。
D 重金属
a 試薬の調製
1) 標準鉛液
特級鉛〔Pb〕1gをトールビーカーに正確にとり、硝酸10mL及び水約30mLを加え加熱して溶かし、冷却後水を加えて正確に1Lとし、標準鉛原液を調製する(この液1mLは、Pbとして1mgを含有する)。使用に際してこの原液の一定量を水で正確に100倍に希釈する(この液1mLは、Pbとして0.01mgを含有する)。
2) 硫化ナトリウム液
硫化ナトリウム5gを水10mL及びグリセリン30mLの混合液に溶かし、しや光したびんに入れて貯蔵する。
b 検液及び比較液の調製
試料10gをネスラー管にとり、水適量を加えて溶かし、40mLとし、これに1mol/L酢酸2mL及び水を加えて50mLとする(この液を検液とする)。
別に標準鉛液1.0mLをネスラー管にとり、1mol/L酢酸2mL及び水を加えて50mLとする(この液を比較液とする)。
c 操作
検液及び比較液に硫化ナトリウム液1滴ずつを加えて混和し、5分間放置した後、両管を白色の背景を用い、上方又は側方から観察して液の色を比較する。
検液の呈する色は、比較液の呈する色より濃いものでないこと。
○ジウレイドイソブタンの試験法
A 純度
a 装置及び器具
1) ガスクロマトグラフ
水素炎イオン化検出器付ガスクロマトグラフ
2) 耐圧フラスコ
250mLの共せん付耐圧フラスコで、せんはスプリングで固定できるもの。
3) カラム
内径 3mm、長さ 3m、ステンレス製又はガラス製
b 試薬
1) 標準ジウレイドイソブタン
ジウレイドイソブタンを約90゚の熱水で2回再結晶を行い、結晶の倍量のメタノールで洗浄し、真空乾燥する。
2) 酢酸エチル
特級酢酸エチルを無水硫酸ナトリウムで脱水後、蒸留した主留分を用いる。
3) pH1.0緩衝液
1mol/L酢酸ナトリウム液200mL、1mol/L塩酸300mL及び水500mLを混合し、pH1.0に調節する。
c ガスクロマトグラフィーの条件
1) カラム充てん剤 ジオクチルフタレート25%セライト545
2) カラム温度 90゚
3) 試料気化室温度 120゚
4) キャリアーガス N2
5) 流速 40mL/min
6) 水素炎 空気1.0kg/cm2 水素70mL/min
d 操作
試料の一定量(ジウレイドイソブタンとして約0.2g)を耐圧フラスコに正確にとり、pH1.0緩衝液100mL及びトルエン20mLを加え、テフロン製マグネット棒を入れ、せんをスプリングで固定する。これを約40゚の水浴に浸し、マグネティックスターラーで20分間激しくかき混ぜた後、氷水中に移し5分間かき混ぜて冷却する。
直ちに内部標準物質として酢酸エチル0.25mLを注射器でとり、針にゴムせんを付け重量を量つた後、冷却した分解液に加え(注射器の重量を量り、その減量から酢酸エチルの採取量を求めておく。)、激しく振り混ぜた後トルエン層12~13mLと無水硫酸ナトリウム4gを遠沈管にとりせんをして遠心機にかけ、分離したトルエン層をガスクロマトグラフィーに供する。
別に標準ジウレイドイソブタン0.15g、0.20g、0.25gをそれぞれ耐圧フラスコに正確にとり、pH1.0緩衝液100mL及びトルエン20mLを加え、以下試料と同様に操作を行う。
e 計算
1) 標準液から得られたガスクロマトグラムから、イソブチルアルデヒドと酢酸エチルとのピーク高比を求め、重量比に対する検量線を作成する。
2) 試料液のガスクロマトグラムから得られたイソブチルアルデヒドと酢酸エチルのピーク高比からその重量比を検量線によって求め、次式によりジウレイドイソブタンの量を算出する。
ジウレイドイソブタンの純度(%)={(重量比×酢酸エチル採取量(g))÷試料採取量(g)}×100
B 水分
試料約5gをひよう量ざらに正確にとり、75±1゚で4時間乾燥し、その減量を水分とする。
C 尿素
a 試料液の調製
試料約5gを500mLのメスフラスコに正確にとり、水約400mLを加え、1分間30~40回転の振り混ぜ機で30分間振り混ぜた後、標線まで水を加え、乾燥ろ紙でろ過する。
b 定量
試料液50mLを蒸留フラスコに正確にとり、メチルレッドを指示薬として0.5%水酸化ナトリウム液で中和し(pH5.6~5.8)、尿素を分解するのに十分な量のウレアーゼを加え、密せんして40~45゚の水浴中に1時間作用させた後冷却する。この分解液に酸化マグネシウム2~3g及び少量のシリコン油を加え、標準硫酸液20mLを正確に入れた受器を接続した水蒸気蒸留装置に連結する。以下粗たん白質の定量法により滴定し、別にウレアーゼの空試験を行い、滴定値を補正した後、尿素性窒素〔N〕の量を求める。これに係数2.1438を乗じて尿素の量とする。
D 重金属
a 試薬の調製
尿素の試験法のDのaによる。
b 検液及び比較液の調製
試料2.0gを石英製又は磁製るつぼにとり、初めは弱く加熱し、次いで強熱しで灰化する。冷後、王水1mLを加え水浴上で蒸発乾固し、残留物を塩酸3滴で潤し、熱湯10mLを加えて2分間加熱する。
次にフェノールフタレインを指示薬とし、10%アンモニア水を液が微赤色となるまで滴加し、1mol/L酢酸2mLを加え、必要があればろ過し、水10mLで洗い、ろ液及び洗液をネスラー管に入れ、水を加えて50mLとする(この液を検液とする)。
別に王水1mLを水浴上で蒸発乾固し、以下検液の調製法と同様に操作し、標準鉛液2.0mL及び水を加えて50mLとする(この液を比較液とする)。
c 操作
検液及び比較液に硫化ナトリウム液1滴ずつを加えて混和し、5分間放置した後、両管を白色の背景を用い、上方又は側方から観察して液の色を比較する。
検液の呈する色は、比較液の呈する色より濃いものでないこと。
イ 尿素又はジウレイドイソブタンを原料とすることができる飼料の種類及びその配合割合は、次の表のとおりとする。
| 原料 | 飼料の種類 | 配合割合 |
|---|---|---|
| 尿素 | 牛用飼料(生後おおむね6月を超えた牛用飼料に限る。) | 2.0%以下 |
| ジウレイドイソブタン | 牛用飼料(搾乳牛以外の生後おおむね6月を超えた牛用飼料に限る。) | 1.5%以下 |
(2) 尿素及びジウレイドイソブタン並びにこれらを含む飼料の製造の方法の基準
ア 尿素
アンモニアと二酸化炭素を高温、高圧で反応させて製造する。この場合、その製造工程中に触媒、固結防止剤その他の物を用いてはならない。
イ ジウレイドイソブタン
尿素とイソブチルアルデヒドを硫酸酸性で反応させて製造する。この場合、その製造工程中に硫酸以外の触媒及び水酸化ナトリウム以外の中和剤を用いてはならない。製品の粒径は、840μmの網ふるいを通過するものでなければならない。
ウ 尿素又はジウレイドイソブタンを原料とする飼料
尿素及びジウレイドイソブタンは、同一飼料の原料として用いてはならない。
(3) 尿素及びジウレイドイソブタンの使用の方法の基準
尿素及びジウレイドイソブタンは、それぞれ単体で使用してはならない。
(4) 尿素及びジウレイドイソブタンの保存の方法の基準
尿素及びジウレイドイソブタンは、湿気の多い場所に保存してはならない。
(5) 尿素及びジウレイドイソブタン並びにこれらを原料とする飼料の表示の基準
ア 尿素若しくはジウレイドイソブタン又はこれらを原料とする飼料には、次に掲げる事項を表示しなければならない。
(ア) 飼料の名称
(イ) 製造(輸入)年月
(ウ) 製造(輸入)業者の氏名又は名称及び住所
(エ) 製造事業場の名称及び所在地(輸入に係るものにあつては、輸入先国名)
イ 尿素及びジウレイドイソブタンには、次に掲げる事項を表示しなければならない。
(ア) 「飼料用」という文字
(イ) 純 度
ウ 尿素又はジウレイドイソブタンを原料とする飼料には、次に掲げる事項を表示しなければならない。
(ア) 対象家畜等
(イ) 尿素又はジウレイドイソブタンの配合割合
(ウ) 使用上の注意事項
(エ) 保存上の注意事項
注
1 使用上の注意事項は、次に掲げる文字(尿素を原料とする飼料にあっては1)から4)まで、ジウレイドイソブタンを原料とする飼料にあっては1)から3)まで)を記載すること。
1) この飼料と他の飼料を併用する場合は、たん白質が過剰とならないよう配慮すること。
2) 新たにこの飼料を給与する場合は、最低3週間の期間をかけて、給与量を徐々に増加させていくこと。
3) 生粕類と混合してこの飼料を給与すると、尿素が急激に分解され、家畜に生理上の障害をきたすおそれがあるので注意すること。
4) 高泌乳牛に給与する場合は、当該乳牛の特性、健康状態等を勘案し、適量の使用を行うよう特に注意すること。
2 保存上の注意事項は、次に掲げる文字を記載すること。
保存に当たつては、吸湿等による品質の低下をきたさないよう配慮すること。
5 動物性油脂又は動物性油脂を原料とする飼料の成分規格及び製造の方法等の基準
(1) 動物性油脂及び動物性油脂を原料とする飼料の成分規格
ア 動物性油脂(獣畜、鳥類又は魚介類を原料として製造された油脂をいい、魚介類のみを原料としてほ乳動物由来たん白質及び家きん由来たん白質(確認済ゼラチン等を除く。)の製造工程と完全に分離された工程において製造されたものを除く。以下同じ。)の不溶性不純物の含有量は、0.15%以下でなければならない。この場合の不溶性不純物の試験法は、次のとおりとする。
試料約20gを精密に量り注1)、特級石油エーテル200mLを加えて溶かした後、重量既知のガラスろ過器(G3注2))でろ過し注3)、ろ過器上の残留物を特級石油エーテル200mLで十分に洗浄する。残留物の入ったガラスろ過器を105±1℃で1時間乾燥し、デシケーター(シリカゲル)で30分間放冷した後、重量を精密に量り、次式により不溶性不純物の含有量を算出する。
不溶性不純物(%)=(W3-W2)÷W1×100
W1:試料採取量(g)
W2:ガラスろ過器の重量(g)
W3:残留物の入ったガラスろ過器の重量(g)
注
1) 試料の油脂は、あらかじめ温湯中で十分溶解し、よくふり混ぜた後、ピペットを用いてビーカーに量りとる。
2) 105±1℃で1時間乾燥し、デシケーター(シリカゲル)で30分間放冷した後、重量を精密に量る。
3) 牛脂の一部には、ろ過中に油脂の一部が固化するものがあるため、秤量後直ちに石油エーテルを加え、完全に溶解した後、10分以内にろ過する。ろ過し難い試料は、吸引ろ過する。
イ ほ乳期子牛等育成用代用乳用配合飼料(ほ乳期子牛等(生後おおむね3月以内の牛、めん羊、山羊及び鹿をいう。)の育成の用に供する配合飼料であって、脱脂粉乳を主原料とするものをいう。以下同じ。)は、動物性油脂(食用の肉から採取した脂肪のみを原料とするものであって、不溶性不純物の含有量が0.02%以下であるもの(以下「特定動物性油脂」という。)を除く。)を含んではならない。
ウ 牛等を対象とする飼料(ほ乳期子牛等育成用代用乳用配合飼料を除く。以下同じ。)は、動物性油脂(牛の脊柱等が混合しないものとして農林水産大臣の確認を受けた工程において製造された油脂(以下「確認済動物性油脂」という。)であつて反すう動物由来動物性油脂(反すう動物に由来する動物性油脂をいい、特定動物性油脂を除く。以下同じ。)を含まないもの並びに特定動物性油脂を除く。)を含んではならない。
エ 家畜等(牛等を除く。)を対象とする飼料は、動物性油脂(確認済動物性油脂及び特定動物性油脂を除く。)を含んではならない。
(2) 動物性油脂及び動物性油脂を原料とする飼料の製造の方法の基準
ア 動物性油脂(特定動物性油脂を除く。)は、ほ乳期子牛等育成用代用乳用配合飼料(ほ乳期子牛等育成用代用乳用配合飼料を製造するための原料又は材料を含む。)に用いてはならない。
イ 動物性油脂(確認済動物性油脂であつて反すう動物由来動物性油脂を含まないもの及び特定動物性油脂を除く。)は、牛等を対象とする飼料に用いてはならない。
ウ 動物性油脂(確認済動物性油脂及び特定動物性油脂を除く。)は、家畜等(牛等を除く。)を対象とする飼料に用いてはならない。
(3) 動物性油脂及び動物性油脂を原料とする飼料の使用の方法の基準
ア 動物性油脂(確認済動物性油脂であつて反すう動物由来動物性油脂を含まないもの及び特定動物性油脂を除く。)を含む飼料は、牛等に対し使用してはならない。
イ 動物性油脂(確認済動物性油脂及び特定動物性油脂を除く。)を含む飼料は、家畜等(牛等を除く。)に対し使用してはならない。
(4) 動物性油脂及び動物性油脂を原料とする飼料の保存の方法の基準
ア 動物性油脂(特定動物性油脂を除く。)を含む飼料は、ほ乳期子牛等育成用代用乳用配合飼料(ほ乳期子牛等育成用代用乳用配合飼料を製造するための原料又は材料を含む。)に混入しないように保存しなければならない。
イ 動物性油脂(確認済動物性油脂であつて反すう動物由来動物性油脂を含まないもの及び特定動物性油脂を除く。)を含む飼料は、牛等を対象とする飼料(飼料を製造するための原料又は材料を含む。)に混入しないように保存しなければならない。
ウ 動物性油脂(確認済動物性油脂及び特定動物性油脂を除く。)を含む飼料は、家畜等(牛等を除く。)を対象とする飼料(飼料を製造するための原料又は材料を含む。)に混入しないように保存しなければならない。
(5) 動物性油脂又は動物性油脂を原料とする飼料の表示の基準
ア 動物性油脂又は動物性油脂を原料とする飼料には、次に掲げる事項を表示しなければならない。
(ア) 飼料の名称
(イ) 製造(輸入)年月
(ウ) 製造(輸入)業者の氏名又は名称及び住所
(エ) 製造事業場の名称及び所在地(輸入に係るものにあつては、輸入先国名)
イ 動物性油脂又は動物性油脂を原料とする粉末油脂(油脂をカゼイン等でコーティングし、粉末にしたものをいう。)には、動物性油脂中の不溶性不純物の含有量を表示しなければならない。
ウ 確認済動物性油脂を含む飼料には、確認済動物性油脂を含む飼料である旨を表示しなければならない。
エ 確認済動物性油脂(反すう動物由来動物性油脂を含むものに限る。)又は特定動物性油脂を含む飼料には、対象家畜等を表示しなければならない。
オ 確認済動物性油脂(反すう動物由来動物性油脂を含むものに限る。)を含む飼料には、次の文字を表示しなければならない。
使用上及び保存上の注意
1 この飼料は、牛、めん羊、山羊及び鹿には使用しないこと(牛、めん羊、山羊又は鹿に使用した場合は処罰の対象となるので注意すること。)。
2 この飼料は、牛、めん羊、山羊及び鹿を対象とする飼料(飼料を製造するための原料又は材料を含む。)に混入しないよう保存すること。
6 食品循環資源又は食品循環資源を原料若しくは材料とする飼料の成分規格及び製造の方法等の基準
(1) 食品循環資源を原料又は材料とする飼料の成分規格
豚を対象とする飼料(飼料を製造するための原料又は材料を除く。以下6において同じ。)は、肉(牛等、豚、いのしし、馬又は家きんに由来するものをいう。以下(1)において同じ。)を扱う事業所等から排出される食品循環資源であつて、肉と接触した可能性があるもの(以下「動物由来食品循環資源」という。)を含んではならない。ただし、次に掲げる動物由来食品循環資源については、この限りでない。
ア 飼料の製造段階で農林水産大臣が定める方法により加熱処理及び製造工程の管理(以下「加熱処理等」という。)が行われたもの(以下「処理済動物由来食品循環資源」という。)
イ 食品の製造段階で農林水産大臣が定める方法により加熱処理等が行われたもの(以下「処理済食品由来動物由来食品循環資源」という。)
ウ 確認済ゼラチン等、確認済豚血粉等、確認済豚肉骨粉等、確認済馬肉骨粉等、確認済原料混合肉骨粉等、確認済チキンミール等、確認済家きん加水分解たん白質等、確認済牛血粉等及び確認済牛肉骨粉等(以下「確認済動物由来たん白質」と総称する。)
(2) 食品循環資源又は食品循環資源を原料若しくは材料とする飼料の製造の方法の基準
ア 食品循環資源
(ア) 豚を対象とする飼料の原料又は材料となる動物由来食品循環資源(処理済動物由来食品循環資源の製造業者に販売されるものを除く。)は、(1)のアの農林水産大臣が定める方法により加熱処理等を行わなければならない。
(イ) 豚を対象とする飼料の原料又は材料となる動物由来食品循環資源は、動物由来食品循環資源(処理済動物由来食品循環資源、処理済食品由来動物由来食品循環資源及び確認済動物由来たん白質を除く。)の製造工程と完全に分離された工程において製造されなければならない。
イ 食品循環資源を原料又は材料とする飼料
(ア) 動物由来食品循環資源(処理済動物由来食品循環資源、処理済食品由来動物由来食品循環資源及び確認済動物由来たん白質を除く。)は、豚を対象とする飼料に用いてはならない。
(イ) 豚を対象とする飼料は、動物由来食品循環資源(処理済動物由来食品循環資源、処理済食品由来動物由来食品循環資源及び確認済動物由来たん白質を除く。)を原料又は材料とする飼料の製造工程と完全に分離された工程において製造されなければならない。
(3) 食品循環資源を原料又は材料とする飼料の使用の方法の基準
動物由来食品循環資源(処理済動物由来食品循環資源、処理済食品由来動物由来食品循環資源及び確認済動物由来たん白質を除く。)を原料又は材料とする飼料は、豚に対し使用してはならない。
(4) 食品循環資源又は食品循環資源を原料若しくは材料とする飼料の保存の方法の基準
ア 動物由来食品循環資源(処理済動物由来食品循環資源、処理済食品由来動物由来食品循環資源及び確認済動物由来たん白質を除く。)を原料又は材料とする飼料は、豚を対象とする飼料に混入しないように保存しなければならない。
イ 動物由来食品循環資源(処理済動物由来食品循環資源、処理済食品由来動物由来食品循環資源及び確認済動物由来たん白質を除く。)は、処理済動物由来食品循環資源、処理済食品由来動物由来食品循環資源及び確認済動物由来たん白質に混入しないように保存しなければならない。
(5) 食品循環資源又は食品循環資源を原料若しくは材料とする飼料の表示の基準
ア 動物由来食品循環資源を原料又は材料とする飼料には、次に掲げる事項を表示しなければならない。
(ア) 飼料の名称
(イ) 製造(輸入)年月
(ウ) 製造(輸入)業者の氏名又は名称及び住所
(エ) 製造事業場の名称及び所在地(輸入に係るものにあつては、輸入先国名)
イ 飼料の原料又は材料となる動物由来食品循環資源(処理済動物由来食品循環資源、処理済食品由来動物由来食品循環資源及び確認済動物由来たん白質を除く。)及び動物由来食品循環資源(処理済動物由来食品循環資源、処理済食品由来動物由来食品循環資源及び確認済動物由来たん白質を除く。)を原料又は材料とする飼料には、対象家畜等を表示しなければならない。
別表第2(第2条関係)
1 飼料添加物一般の通則
(1) 飼料添加物の適否は、各飼料添加物の成分規格及び製造の方法等の基準(以下「各条」という。)の規定、飼料添加物一般の通則及び飼料添加物一般の試験法(以下「一般試験法」という。)の規定により判定する。ただし、物理的・化学的性質の項の臭い、味、結晶形、溶解性、液性、安定性、吸光度、凝固点、屈折率、旋光度、粘度、比重及び融点は、参考に供したもので、適否の判断基準を示すものではない。なお、飼料添加物の有用性又は安定性を高めるため、各条に規定する製剤に安定剤、滑沢剤、結合剤、湿潤剤、乳化剤、被覆剤、分散剤、崩壊剤、保存剤又は溶解補助剤を用いることができる。
(2) 物質名の次に( )で分子式を付けたものは、化学的純物質を意味する。
(3) 主な計量の単位については、次の記号を用いる。
| メートル | m | センチメートル | cm | |
| ミリメートル | mm | マイクロメートル | μm | |
| ナノメートル | nm | 平方センチメートル | cm2 | |
| リットル | L | ミリリットル | mL | |
| マイクロリットル | μL | トン | t(1,000kg) | |
| キログラム | kg | グラム | g | |
| ミリグラム | mg | マイクログラム | μg | |
| キロパスカル | kPa | モル | mol | |
| マイクロモル | μmol | モル毎リットル | mol/L | |
| セルシウス度 | ℃ |
(4) 質量百分率を示すには、%の記号を用いる。ただし、溶液100mL中の物質含量(g)を示すにはw/v%、溶液100mL中の物質含量(mL)を示すにはv/v%、溶液100g中の物質含量(mL)を示すにはv/w%の記号を用いる。
(5) 抗生物質及び酵素の量は、抗生物質にあっては当該抗生物質の力価で、酵素にあっては当該酵素の酵素力単位で示す。
(6) 標準温度は20℃、常温は15~25℃、室温は1~30℃、微温は30~40℃とする。冷所は、別に規定する場合を除き、15℃以下の場所とする。冷水は10℃以下、微温湯は30~40℃、温湯は60~70℃、熱湯は約100℃の水とする。水浴上又は水浴中で加熱するとは、別に規定する場合を除き、沸騰した水浴又は約100℃の蒸気浴を用いて加熱することをいう。
(7) 飼料添加物の試験に用いる水は、別に規定する場合を除き、精製水とする。
(8) 滴数を量る際には、20℃において、精製水20滴を滴下するとき、その質量が0.90~1.10gとなるような器具を用いる。
(9) 数値を整理して小数点以下n桁とする場合は、(n+1)桁目の数値を、四捨五入する。
(10) 原子量は、2007年国際原子量表によるものとする。分子量は、この表により計算した後、小数点以下2桁までに整理する。
(11) 減圧は、別に規定する場合を除き、2.0kPa以下とする。
(12) 液性を酸性、アルカリ性又は中性として示す場合は、別に規定する場合を除き、リトマス紙を用いて試験する。液性を詳しく示すには、pH値を用いる。
(13) 溶質名の次に「溶液」と記載し、特にその溶媒名を示さないものは、水溶液を示す。
(14) 溶液の濃度を「(1→3)」、「(1→10)」、「(1→100)」等と記載したものは、固体は1g、液体は1mLを溶媒に溶かし、全量をそれぞれ3mL、10mL、100mL等とした場合の割合を示す。また、混液を「(1:10)」、「(5:3:1)」等で示したものは、液体の1容量と10容量の混液、5容量と3容量と1容量の混液等を示す。
(15) 飼料添加物の試験は、別に規定する場合を除き、常温で行い、操作直後に観察するものとする。ただし、温度の影響のあるものの判定は、標準温度における状態を基準とする。
(16) 物理的・化学的性質の項において、「白色」と記載したものは白色又はほとんど白色、「無色」と記載したものは無色又はほとんど無色を示すものとする。色調を試験するためには、別に規定する場合を除き、固体の飼料添加物にあっては当該飼料添加物1gを白紙上又は白紙上に置いた時計皿にとり観察し、液体の飼料添加物にあっては内径15mmの無色の試験管に入れ白色の背景を用い液層を30mmとして観察する。液体の飼料添加物の澄明性を試験するには、黒色又は白色の背景を用い、前記の方法を準用する。液体の飼料添加物の蛍光を観察するには、黒色の背景を用い、白色の背景は用いない。
(17) 物理的・化学的性質の項において、「無臭又は臭いがない」と記載したものは、臭いがない、又はほとんど臭いがないことを示すものである。臭いを試験するためには、別に規定する場合を除き、固体又は液体の飼料添加物は、その1gを100mLのビーカーにとり、行う。
(18) 物理的・化学的性質の項において、溶解性を示す用語は、次によるものとする。溶解性は、別に規定する場合を除き、飼料添加物を、固形の場合は粉末とした後、溶媒中に入れ、20±5℃で5分間ごとに30秒間強く振り混ぜるとき、30分以内に溶ける度合いをいう。
| 用語 | 溶質1g又は1mLを溶かすために要する溶媒量 | |
|---|---|---|
| 極めて溶けやすい | 1mL未満 | |
| 溶けやすい | 1mL以上 | 10mL未満 |
| やや溶けやすい | 10mL以上 | 30mL未満 |
| やや溶けにくい | 30mL以上 | 100mL未満 |
| 溶けにくい | 100mL以上 | 1,000mL未満 |
| 極めて溶けにくい | 1,000mL以上 | 10,000mL未満 |
| ほとんど溶けない | 10,000mL以上 | |
(19) 飼料添加物の試験において、飼料添加物が溶媒に溶ける又は混和するとは、澄明に溶ける、又は澄明に混和することをいう。ただし、僅かの繊維又はごみを認めても差し支えない。
(20) 確認試験は、飼料添加物又は飼料添加物中に含有されている主成分等を確認するために行う試験とする。
(21) 純度試験は、飼料添加物中の混在物を試験するために行うもので、各条の他の試験項目と共に飼料添加物の純度を規定する試験とし、通例、その混在物の種類及びその量の限度を規定する。この試験の対象となる混在物は、飼料添加物を製造する若しくは保存する間に混在を予想されるもの又は有害な混在物、例えば、重金属、ヒ素等とする。また、異物を用い、又は加えることが予想される場合についても、この試験を行う。
(22) 「澄明」、「ほとんど澄明」、「僅かに微濁」、「微濁」又は「混濁」と記載したものは、次の基準によるものとする。
濁度標準原液 0.1mol/L塩酸14.1mLに水を加えて50mLとする。この液1mLは、C11mgを含む。
濁度標準液 濁度標準原液10mLに水を加えて1,000mLとする。この液1mLは、C1 0.01mgを含む。
ア 澄明 濁度標準液0.2mLに水を加えて20mLとし、これに硝酸(1→3)1mL、2w/v%デキストリン溶液0.2mL及び2w/v%硝酸銀溶液1mLを加え、15分間放置したときの濁度以下とする。ただし、浮遊物等の異物の混入をほとんど認めないものでなければならない。
ほとんど澄明 濁度標準液0.5mLに水を加えて20mLとし、これに硝酸(1→3)1mL、2w/v%デキストリン溶液0.2mL及び2w/v%硝酸銀溶液1mLを加え、15分間放置したときの濁度とする。ただし、浮遊物等の異物の混入をほとんど認めないものでなければならない。
ウ 僅かに微濁 濁度標準液1.2mLに水を加えて20mLとし、これに硝酸(1→3)1mL、2w/v%デキストリン溶液0.2mL及び2w/v%硝酸銀溶液1mLを加え、15分間放置したときの濁度とする。
エ 微濁 濁度標準液6mLに水を加えて20mLとし、これに硝酸(1→3)1mL、2w/v%デキストリン溶液0.2mL及び2w/v%硝酸銀溶液1mLを加え、15分間放置したときの濁度とする。
オ 混濁 濁度標準原液0.3mLに水を加えて20mLとし、これに硝酸(1→3)1mL、2w/v%デキストリン溶液0.2mL及び2w/v%硝酸銀溶液1mLを加え、15分間放置したときの濁度とする。
(23) 乾燥又は強熱するときの恒量とは、別に規定する場合を除き、引き続き更に1時間乾燥又は強熱するときの前後の秤量差が、前回に量った乾燥物又は強熱した残留物の質量の0.10%以下であることをいう。ただし、化学はかりを用いたときの秤量差が0.5mg以下の場合、セミミクロ化学はかりを用いたときの秤量差が0.05mg以下の場合及びミクロ化学はかりを用いたときの秤量差が0.005mg以下の場合は、恒量とみなす。
(24) 定量法は、飼料添加物の組成、成分の含量又は含有単位等を物理的、化学的又は生物学的方法により測定する試験法である。
(25) 定量に供する試料又は標準品の採取量に「約」を付けたものは、記載された量の±10%の範囲をいう。また、試料又は標準品について単に「乾燥し」とあるのは、各条又は標準品の乾燥減量の項と同じ条件で乾燥することとし、試料について単に「強熱し」とあるのは、各条の強熱減量の項と同じ条件で強熱することとする。
(26) 各条の定量法で得られる成分含量の値について、単にある%以上を示し、その上限を示さない場合は、101.0%を上限とする。また、含量の項において、例えば、「表示量の90~110%に対応する純品を含む。」と規定してあるのは、化学的純物質又はこれに対応するものを100%含むように調製し、これを定量するとき、上記の範囲内にあることを示し、また、「表示力価の85~125%を含む。」と規定してあるのは、有効期間中表示力価を保つように調製し、これを定量するとき、上記の範囲内にあることを示す。
(27) 一般試験法及び各条に規定する試験法に代わる方法で、規定の方法以上の正確さ及び精密さがある場合は、その方法を用いることができる。ただし、その結果について疑いのある場合は、規定の方法で最終の判定を行う。
(28) 容器とは、飼料添加物を入れるもので、栓、蓋等容器の構成の一部として用いるものも含む。
(29) 密閉容器とは、日常の取扱い又は通常の保存状態において、固体の異物が混入することを防ぎ、内容飼料添加物が損失しないように保護することができる容器をいう。密閉容器の規定がある場合には、気密容器又は密封容器を用いることができる。
(30) 気密容器とは、日常の取扱い又は通常の保存状態において、液体若しくは固体の異物又は水分が浸入せず、内容飼料添加物が損失し、風解し、潮解し、又は蒸発しないように保護することができる容器をいう。気密容器の規定がある場合には、密封容器を用いることができる。
(31) 密封容器とは、日常の取扱い又は通常の保存状態において、気体又は微生物の侵入するおそれのない容器をいう。
(32) 遮光した容器とは、光の透過を防ぐ容器又は光の透過を防ぐ包装を施した容器をいう。
2 飼料添加物一般の成分規格
組換えDNA技術により得られた生物を利用して飼料添加物を製造する場合は、当該飼料添加物は、その安全性につき、農林水産大臣の定めるところにより、農林水産大臣の確認を受けたものでなければならない。ただし、当該飼料添加物が安全性の確保に支障がないものとして農林水産大臣が定める基準に適合する場合は、この限りでない。
3 飼料添加物一般の製造の方法の基準
(1) ア 成分についての規格が定められた飼料添加物を原料とする場合は、当該規格に適合するもの(法第5条第1項の検定を要するものにあっては、当該検定に合格したものに限る。)を用いなければならない。
イ 成分についての規格に適合しない製造用原体を原料とする製剤は、飼料添加物の原料としてはならない。
(2) 別表第1の1の(2)のウの表の同一欄内の2以上の飼料添加物を用いて飼料添加物を製造してはならない。
(3) 2以上の飼料添加物を用いて飼料添加物を製造する場合は、それぞれの飼料添加物の効果が阻害されないようにし、かつ、それぞれの飼料添加物の成分の定量試験、確認試験その他の試験が困難とならないようにしなければならない。
(4) 液状の飼料添加物は、各条に規定されているもの以外は製造してはならない。
(5) 賦形物質、希釈物質その他の飼料添加物の製造に用いる物は、次に掲げる要件の全てを満たすものでなければならない。
ア 有害な物質を含まず、若しくは病原微生物により汚染されず、又はこれらの疑いがないこと。
イ 当該飼料添加物の効果を阻害しないこと。
ウ 当該飼料添加物の成分の定量試験、確認試験その他の試験が困難とならないこと。
エ リグノスルホン酸カルシウム及びリグノスルホン酸ナトリウムは、次に掲げる要件の全てを満たすものであること。
(ア) リグノスルホン酸カルシウム 木材からパルプを製造する際に得られるパルプ液であって、亜硫酸水素カルシウムを加えたものを加圧蒸煮し、かつ、乾燥させることにより得られる褐色の粉末
スルホン酸イオウ 5.0%以上
カルシウム 7.0%以下
50%溶液の粘度 3,000センチポアズ以下
鉛 1mg/kg以下
還元糖 30.0%以下
乾燥減量 10.0%以下
強熱残分 20.0%以下
(イ) リグノスルホン酸ナトリウム 木材からパルプを製造する際に得られるパルプ液であって、亜硫酸水素ナトリウムを加えたものを加圧蒸煮し、かつ、乾燥させることにより得られる褐色の粉末
スルホン酸イオウ 5.0%以上
ナトリウム 10.0%以下
50%溶液の粘度 3,000センチポアズ以下
鉛 1mg/kg以下
還元糖 30.0%以下
乾燥減量 10.0%以下
強熱残分 20.0%以下
(6) 賦形物質及び希釈物質(以下「賦形物質等」という。)は、次に掲げる物を用い、その他の飼料添加物の製造に用いる物は、各条に規定されているものであること。ただし、液状の飼料添加物は、各条に規定されているもの以外は使用してはならない。 アラビアゴム、アルブミン、エチルセルロース、カオリン、カゼイン、活性グルテン、カラゲーナン、カラメル、カルナウバろう、含水二酸化ケイ素、含水無晶形酸化ケイ素、肝臓粉末、寒天、キサンタンガム、キトサン、きな粉、グァーガム、グリセリン、グルコマンナン、グルテン、グルテンミール、ケイ酸、ケイ酸カルシウム、ケイ酸マグネシウム、軽質無水ケイ酸、軽質流動パラフィン、ケイソウ土、硬化油、高級飽和脂肪酸、小麦粉、小麦ミドリング、米ぬか、米ぬか油かす、コーングリッツ、コーングルテンフィード、コーンコブミール、コーンスターチ、シイタケホダ木粉末、ジスチラーズグレイン、ジスチラーズグレインソリュブル、脂肪酸、脂肪酸カルシウム、食塩、植物性油脂、ステアリン酸カルシウム、ゼオライト、ゼラチン、セルロース、ソイビーンミルラン、ソルビトール、脱脂魚粉、脱脂粉乳、炭酸カルシウム、大豆油かす、大豆皮、大豆粉、タマリンド種子多糖類、タルク、炭酸ナトリウム、デキストラン、デキストリン、天然ケイ酸アルミニウム、デンプン、α-デンプン、動物性油脂、トウモロコシ粉、トラカントガム、トルラ酵母、乳糖、濃縮大豆たん白、麦芽糖、白糖、バーミキュライト、パン酵母、ビール酵母、ファーセレラン、ふすま、ブドウ糖、プルラン、ペクチン、変性食用デンプン、ベントナイト、ポテトパルプ、ホワイトフィッシュミール、D-マンニトール、無水ケイ酸、無水ケイ酸塩類、もみがら、もみがら粉末、リグノスルホン酸カルシウム、リグノスルホン酸ナトリウム、流動パラフィン、リン酸一水素カルシウム、リン酸三カルシウム、リン酸二水素カルシウム、レシチン、ローカストビーンガム
(7) 2以上の原料又は材料を用いる場合には、これらを原料又は材料として製造する飼料添加物が、均質なものとなるようにしなければならない。
(8) 組換えDNA技術により得られた微生物を利用して飼料添加物を製造する場合は、農林水産大臣が定める基準に適合する旨の農林水産大臣の確認を得た方法で製造しなければならない。
4 飼料添加物一般の保存の方法の基準
(1) 有害な物質を含み、若しくは病原微生物により汚染され、又はこれらの疑いがある場所に保管してはならない。また、有害な物質を含み、若しくは病原微生物により汚染され、又はこれらの疑いがある容器若しくは包装材料を用いて保存してはならない。
(2) 表示の基準に基づき保存上の注意事項が表示されている飼料添加物は、当該保存上の注意事項を遵守して保存しなければならない。
5 飼料添加物一般の表示の基準
(1) 輸出用又は試験研究用の飼料添加物には、「輸出用」又は「試験研究用」の文字を表示しなければならない。
(2) 飼料添加物には、次に掲げる事項を表示しなければならない。
ア 飼料添加物の名称(一般名又は商品名)
イ 「飼料添加物」の文字
ウ 製造番号又は製造記号
エ 製造(輸入)業者又は販売業者の氏名又は名称及び住所
オ 製造事業場の名称及び所在地(輸入に係るものにあっては、輸入先国名及び製造業者名)
カ 有効成分名及び含量並びに賦形物質等の名称(ただし、着香料にあっては、有効成分名及び含量の表示を要しない。また、各条に定量法の定められていない飼料添加物(着香料を除く。)の含量にあっては、製造用原体の含有率を重量パーセントで表示するものとする。)
キ 製造(輸入)年月日及び有効期間(ただし、有効期間の表示にあっては、各条において定められているものに限る。)
ク 用いることができる飼料の種類及び量
ケ 保存上の注意事項
注
1 保存上の注意事項には、当該飼料添加物について定められた保存の方法の基準に従い保存するべき旨記載すること。
2 飼料又は飼料添加物の製造業者のみに販売する場合には、農林水産大臣の承認を受けて「製造業者専用」の文字を表示し、上記の表示すべき事項の一部の表示を省略することができる。
(3) 表示は、法第32条第1項の規定に基づく表示の基準に従い行う表示に準じて行うものとする。
6 飼料添加物一般の試験法
一般試験法は、共通の試験法及びこれに関連する事項をまとめたものである。別に規定する場合を除き、液体クロマトグラフ法、塩化物試験法、炎色反応試験法、ガスクロマトグラフ法、乾燥減量試験法、吸光度測定法、凝固点測定法、強熱減量試験法、強熱残分試験法、屈折率測定法、原子吸光光度法、抗菌活性試験法、抗生物質の力価試験法、酵素力試験法、1,4-ジオキサン試験法、重金属試験法、水分定量法、生菌剤試験法、生菌剤定量法、赤外吸収スペクトル測定法、旋光度測定法、粗脂肪定量法、粗繊維定量法、窒素定量法、定性反応、鉛試験法、バイオオートグラフ法、薄層クロマトグラフ法、pH測定法、比重測定法、ヒ素試験法、ビタミンA定量法、ビタミンD定量法、沸点測定法及び蒸留試験法、融点測定法、誘導結合プラズマ発光分光分析法及び誘導結合プラズマ質量分析法、硫酸塩試験法、硫酸呈色物試験法並びにろ紙クロマトグラフ法は、それぞれ規定するところにより行う。
(1) 液体クロマトグラフ法
液体クロマトグラフ法は、固定相として適当な充填剤を詰めたカラム中に、移動相として液体をポンプ等で加圧して流すことにより、カラムに注入された混合物を固定相に対する保持力の差を利用してそれぞれの成分に分離し、分析する方法であって、液体試料又は溶液にできる試料に適用でき、物質の確認、純度の試験又は定量等に用いる。
カラムに注入された混合物は、各成分に固有の比率で、移動相と固定相とに分布する。この比を質量分布比k′という。
k′=固定相に存在する量/移動相に存在する量
質量分布比と保持時間tR(試料注入時からピークの頂点が溶出されるまでの時間)との間には、次の関係があるので、同一カラムについては、温度と移動相の組成及び流量が一定の場合、保持時間は物質に固有の値となる。
tR=(1+k′)t0
t0:k′=0の物質の試料注入時からピークの頂点までの時間
装置
通例、移動相送液用ポンプ、試料導入部、カラム、検出器及び記録装置からなり、必要に応じてカラムは、恒温槽により恒温に保たれる。ポンプは、カラム及び連結チューブの中を一定量で移動相を送液できるものとする。カラムは、液体クロマトグラフ法用に調製した粒径が3~50μmの一定の大きさに揃った充填剤を、内径2~8mm、長さ10~100cmの管に均一に充填したものとする。なお、別に規定するものを除き、次式で定義される分離度RSを各条に規定する。
RS=2(tR1-tR2)/1.67(Wh1+Wh2)
tR1,tR2:分離度測定を用いる2つの物質の保持時間
Wh1,Wh2:各ピークのピーク高さの中点におけるピーク幅
検出器は、通例、紫外及び可視の吸光光度計、示差屈折計、蛍光光度計等移動相とは異なる試料の性質を検出するものであり、数μg以下の試料に対して濃度に比例した信号を出すものとする。検出器により得られる信号の強さは記録装置により記録される。
操作法
装置をあらかじめ調整した後、各条に規定する条件で検出器、カラム及び移動相を用い、移動相を一定流量で流してカラムを規定の温度で平衡にした後、各条に規定する方法で調製した試料溶液をマイクロシリンジ又は試料バルブを用いて試料注入部から注入する。分離された成分を検出器により検出し、記録装置を用いてクロマトグラムとして記録する。試料の確認は、保持時間が一致すること又は標準試料を添加してピークの幅が広がらないことにより行う。定量は、通例、内部標準法によるが、適当な内部標準物質が得られない場合は、絶対検量線法によるものとする。
① 内部標準法
被検成分にできる限り近い保持時間を有し、いずれのピークとも完全に分離する化学的に安定な物質を内部標準物質として選び、その一定量に対して標準被検成分を段階的に加えた標準液を数種類調製する。この一定量ずつを注入して得られたクロマトグラムから、被検物質のピーク高さ又はピーク面積と内部標準物質のピーク高さ又はピーク面積との比を求める。この比を縦軸に、標準被検成分の量を横軸にとり、検量線を作成する。この検量線は、通例、原点を通る直線となる。次に、同量の内部標準物質を加えた試料溶液を調製し、検量線を作成したときと同一条件でクロマトグラムを記録し、被検成分のピーク高さ又はピーク面積と、内部標準物質のピーク高さ又はピーク面積との比を求め、検量線を用いて定量を行う。
② 絶対検量線法
標準被検成分を段階的にとり、標準液を調製し、この一定量ずつを正確に注入する。得られたクロマトグラムから縦軸に標準被検成分のピーク高さ又はピーク面積、横軸に標準被検成分量をとり、検量線を作成する。この検量線は、通例、原点を通る直線となる。次に、各条に規定する方法で試料溶液を調製し、検量線を作成したときと同一条件でクロマトグラムを記録し、被検成分のピーク高さ又はピーク面積を測定し、検量線を用いて定量を行う。
ピーク測定は通例、次のいずれかの方法による。
① ピーク高さ法
ピークの頂点からベースラインへ下ろした垂線とピークの両すそを結ぶ接線との交点から頂点までの長さを測定する。
② ピーク面積法
(ⅰ) 半値幅法 ピーク高さの中点におけるピーク幅にピーク高さを乗じる。
(ⅱ) 自動面積測定法 デジタルインテグレーター等を用いて面積を測定する。
(2) 塩化物試験法
塩化物試験法は、試料中に混在する塩化物の限度試験とする。
各条には、塩化物(Clとして)の限度を( )内に付記する。
操作法
別に規定する場合を除き、次の方法によるものとする。。
各条に規定する量の試料をネスラー管に入れ、適量の水を加えて溶かし、40mLとする。これに希硝酸6mL及び水を加え、50mLとし、試料溶液とする。別に、各条に規定する量の0.01mol/L塩酸を量り、希硝酸6mL及び水を加え、50mLとし、比較液とする。この場合、試料溶液が澄明でないときは、両液を同条件でろ過する。
試料溶液及び比較液にそれぞれ硝酸銀試液1mLを加え、混和し、直射日光を避け、5分間放置した後、黒色の背景を用い、ネスラー管の上方又は側方から観察して混濁を比較する。
試料溶液の呈する混濁は、比較液の呈する混濁より濃くてはならない。
(3) 炎色反応試験法
炎色反応試験法は、ある種の金属塩が鋭敏にブンゼンバーナーの無色炎をそれぞれ固有の色に染める性質を利用して、その金属塩の定性を行う方法である。
操作法
炎色反応の試験に用いる白金線は、径約0.8mmで、先端は直線のままで用いる。試料が固体の場合は、塩酸少量を加えてかゆ状とし、その少量を白金線の先端約5mmの部分につけ、水平に保って無色炎中に入れ、試験する。また、試料が液体の場合は、白金線の先端を試料中に約5mm浸し、静かに引き上げて、以下固体の場合と同様に試験する。
炎色反応が持続するとは、その反応が約4秒間持続することをいう。
(4) ガスクロマトグラフ法
ガスクロマトグラフ法は、適当な固定相を用いて作られた分離管内を、移動相に気体(キャリヤーガス)を用い、混合物を気体状態で展開させて、それぞれの成分に分離する方法であって、気体試料又は気化し得る液体若しくは固体試料に適用でき、物質の確認、純度の試験又は定量等に用いる。
固定相に適当な粒度の吸着性担体を用いる場合を気-固クロマトグラフ法といい、適当な粒度の不活性担体を液相で被覆したもの又は毛細管の内壁を液相で被覆したものを用いる場合を気-液クロマトグラフ法という。
装置
通例、キャリヤーガス送入部、試料送入部、恒温槽に内蔵された分離管、検出器及び記録計からなる。
操作法
別に規定する場合を除き、次の方法によるものとする。
装置をあらかじめ調整した後、各条に規定する条件で分離管、検出器、温度及びキャリヤーガス流量を設定し、各条に規定する量の試料溶液又は標準液をガスクロマトグラフ用マイクロシリンジを用いて試料送入部から注入し、分離された成分を検出器により検出し、記録計を用いてクロマトグラムを作成する。
クロマトグラム上の成分のピーク位置は、保持時間(試料溶液を注入してから成分のピークの頂点が現れるまでの時間)又は保持容量(保持時間×キャリヤーガス流量)で表し、これらは、一定条件では物質に特有の値を示す。これにより試料成分の確認を行う。
また、クロマトグラム上の成分のピーク面積、ピーク高さ等から試料成分の定量を行う。
定量は、通例、次のいずれかの方法によるものとする。
① 内部標準法
各条に規定する内部標準物質の一定量に対して、標準被検成分の既知量をそれぞれ段階的に加えて標準液を調製し、この一定量ずつを注入する。クロマトグラムから縦軸に標準被検成分のピーク面積又はピーク高さと内部標準物質のピーク面積又はピーク高さとの比をとり、横軸に標準被検成分量と内部標準物質量との比又は標準被検成分量をとって検量線を作成する。
次に、各条に規定する方法で試料溶液を調製する。ただし、試料溶液の調製には、あらかじめ標準液の場合と同量の内部標準物質を加える。検量線を作成したときと同一条件で得たクロマトグラムから被検成分のピーク面積又はピーク高さと内部標準物質のピーク面積又はピーク高さとの比を求め、検量線から被検成分量を求める。
内部標準物質として、そのピークが被検成分のピーク位置にできる限り近く、被検成分以外のピークとも完全に分離する化学的に安定な物質を用いる。
② 絶対検量線法
標準被検成分を段階的にとり、標準液を調製し、この一定量ずつを注入する。クロマトグラムから縦軸に標準被検成分のピーク面積又はピーク高さ、横軸に標準被検成分量をとり、検量線を作成する。次に、各条に規定する方法で試料溶液を調製する。検量線を作成したときと同一条件でクロマトグラムを作成し、検量線から被検成分量を求める。この方法は、全測定操作を厳密に一定に保って行う必要がある。
③ 面積百分率法
クロマトグラムから得られた各成分のピーク面積の総和を100とし、それに対するそれぞれの成分のピーク面積の比から組成比を求める。ただし、正確な定量値を得るためには、検出器の感度に基づく各成分のピーク面積の補正を行う必要がある。
ピーク測定は通例、次のいずれかの方法による。
① ピーク高さ法
ピークの頂点からベースラインへ下ろした垂線とピークの両すそを結ぶ接線との交点から頂点までの長さを測定する。
② ピーク面積法
(ⅰ) 半値幅法 ピーク高さの中点におけるピーク幅にピーク高さを乗じる。
(ⅱ) 自動面積測定法 デジタルインテグレーター等を用いて面積を測定する。
(5) 乾燥減量試験法
乾燥減量試験法は、試料を各条に規定する条件で乾燥し、その減量を測定する方法である。この方法は、乾燥することにより失われる試料中の水分、結晶水の全部又は一部及び揮発性物質等の量を測定するために用いる。
各条に、例えば、「1.0%以下(1g,105℃,4時間)」と規定するものは、本品約1gを0.1mgの桁まで量り、その数値を記録し、105℃で4時間乾燥するとき、その減量が本品1gにつき、10mg以下であることを示す。また、「0.5%以下(1g,減圧,五酸化リン,4時間)」と規定するものは、本品約1gを0.1mgの桁まで量り、その数値を記録し、五酸化リンを乾燥剤としたデシケーターに入れ、4時間減圧乾燥するとき、その減量が本品1gにつき、5mg以下であることを示す。
操作法
はかり瓶をあらかじめ各条に規定する方法に準じて30分間乾燥し、その質量を0.1mgの桁まで量り、その数値を記録する。試料は、各条に規定する量の±10%の範囲内で採取し、はかり瓶に入れ、別に規定する場合を除き、その層が5mm以下になるように広げた後、その質量を0.1mgの桁まで量り、その数値を記録し、これを乾燥器に入れ、各条に規定する条件で乾燥する。試料が大きいときは、手早く粉砕して径2mm以下としたものを用いる。乾燥した後、乾燥器から取り出し、質量を0.1mgの桁まで量り、その数値を記録する。加熱した場合は、デシケーター(シリカゲル)で放冷し、質量を0.1mgの桁まで量り、その数値を記録する。加熱温度は、各条に規定する温度の±2℃の範囲とする。各条に規定する乾燥温度よりも低温で融解する試料は、融解温度より5~10℃低い温度で、1~2時間乾燥した後、各条に規定する条件で乾燥する。乾燥剤は、各条に規定するものを用い、しばしば取りかえる。
(6) 吸光度測定法
吸光度測定法は、物質が一定の狭い波長範囲の光を吸収する度合いを測定する方法である。単色光がある物質の溶液を通過するとき、透過光の強さ(I)の入射光の強さ(I0)に対する比率を透過度(t)といい、これを百分率で表したものを透過率(T)という。また、透過度の逆数の常用対数を吸光度(A)という。
t=I/I0
T=(I/I0)×100=100t
A=log(I0/I)=-logt=2-logT
物質の溶液に光を通すとき、吸光度は、その光の波長により異なる。したがって、少しずつ波長の異なった光について吸光度を測定し、これらの吸光度と波長との関係を示す曲線を描くと吸収スペクトルが得られる。吸収スペクトルから、その物質の吸収の極大波長(λmax)及び極小波長(λmin)を知ることができる。
吸収スペクトルは、その物質の化学構造により定まることから、吸収の極大波長若しくは極小波長を測定し、又は特定の2つの波長における吸光度の比を測定することにより、確認又は純度の試験を行う。また、通例、極大波長における一定濃度の溶液の吸光度を測定することにより、定量を行う。
吸光度(A)は、溶液の濃度(c)及び層長(l)に比例する。A=kcl
lを1cm、cを1w/v%溶液に換算したときの吸光度を比吸光度(E1%1cm)、l を1cm、cを1molの溶液に換算したときの吸光度をモル吸光係数(ε)という。吸収の極大波長におけるモル吸光係数は、εmaxで表わす。
E1%1cm=A/(c(%)×l) ε=A/(c(mol)×l)
l:層長(cm)
A:吸光度
c(%):溶液の濃度(w/v%)
c(mol):溶液のモル濃度(mol/L)
装置
測定装置として光電分光光度計を用いる。光電分光光度計は、分光装置及び光電光度計を備えたものであり、光源としては、可視部の測定にあってはタングステンランプを、紫外部の測定にあっては水素放電管又は重水素放電管を用いる。紫外部の吸収測定にあっては石英製のセルを、可視部の吸収測定にあってはガラス製又は石英製のセルを用いる。なお、別に規定する場合を除き、層長は、1cmとする。
操作法
通例、まず波長目盛りを規定する測定波長に合わせ、暗電流をゼロに調整した後、対照液を入れたセルを光路に入れ、シャッターを開き、吸光度がゼロを示すように調整する。対照液は、別に規定する場合を除き、試験に用いた溶媒を用いる。次に、測定しようとする溶液を入れたセルを光路に入れかえ、このとき示す吸光度を読み取る。特に波長幅を規定する場合は、それにより測定を行う。
紫外部の吸収測定に用いる溶媒の吸収については、特に考慮し、測定の妨げにならないものを用いる。なお、波長及び吸光度目盛りの補正は、次の方法によるものとする。
波長目盛りは、通例、石英水銀アーク灯若しくはガラス水銀アーク灯による239.95nm、253.65nm、302.15nm、313.16nm、334.15nm、365.48nm、404.66nm、435.83nm若しくは546.10nm、水素放電管による486.13nm若しくは656.28nm又は重水素放電管による486.02nm若しくは656.10nmの線を用いて補正する。
吸光度目盛りは、重クロム酸カリウム(標準試薬)を0.005mol/L硫酸に溶かし、0.006w/v%とした溶液を用いて補正する。この溶液のE1%1cmは波長235nm(極小)、257nm(極大)、313nm(極小)及び350nm(極大)において、それぞれ125.2、145.6、48.9及び107.0とする。
(7) 凝固点測定法
凝固点とは、次の方法で操作したとき、一定になったときの温度をいう。
装置
図に示すものを用いる。
| A:空気外とう ガラス製で、内外の両壁に曇り止めシリコーン油を塗る。 | 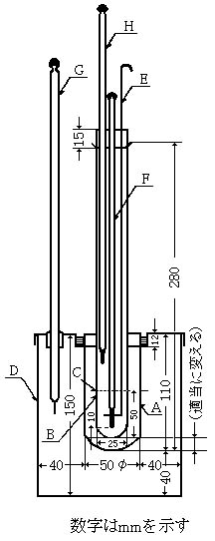 |
| B:試料容器 硬質ガラス製試験管で、管の両壁に曇り止めシリコーン油を塗る。ただし、試料に接する部分には塗らない。 空気外とう中に差し込み、コルク栓で固定する。 | |
| C:標線 | |
| D:浴槽 ガラス製のものとする。 | |
| E:かき混ぜ棒 ガラス製で、径3mm、下端を外径18mmの輪状にしたものとする。 | |
| F:温度計 4号~6号 | |
| G:温度計 1号又は2号 | |
| H:補助温度計 温度計1号を用い、その水銀球の位置が試料の上面と温度計Fの示度(凝固点)との中央部となるようにする。 |
浴液
加熱浴液 水、グリセリン、リン酸トリクレシル、植物性油脂、硫酸等適当なものとする。
常温で試料が固体の場合は、図とは別に、加熱浴槽を設ける。形状、材質等は、適当なものとする。
冷却浴液 凝固点に応じて、次のものを用いる。
80℃以上の場合:グリセリン、リン酸トリクレシル、植物性油脂等適当なものとする。
80~0℃の場合:水又は氷
0~-10℃の場合:砕氷及び塩化ナトリウム、又はメチルアルコール及びドライアイス
操作法
① 試料が常温で固体の場合
清浄かつ乾燥した試料容器Bに、乾燥試料を、溶融したときに液面が標線Cに一致するように入れ、予想する凝固点より20℃以上高くならないように加熱浴中で注意しながら加熱する。完全に試料を溶融させた後、浴中から取り出し、図に示すように温度計F、補助温度計H及びかき混ぜ棒を挿入し、コルク栓で固定し、さらに、予想する凝固点よりも5℃低い温度の浴液を入れた冷却浴に浸す。
かき混ぜ棒を上下に動かし、試料の温度が予想する凝固点よりも5℃高い温度に達したとき、かき混ぜ棒を毎分20~30回の割合で穏やかに上下に動かし、30秒間ごとに温度を読む。
温度は初めは緩やかに降下するが、結晶を生じて上昇し始めたら、かき混ぜるのを止め、10秒間ごとに温度を読み、1分間一定に止まったときの温度計F及び補助温度計Hの温度を読み取り、次式により凝固点を算出する。
T=t+0.00016(t-t′)n
T:凝固点(℃)
t:温度計の示度(℃)
t′:補助温度計の示度(℃)
n:温度計水銀線の液外にある度数(℃)
② 試料が常温で液体の場合
清浄かつ乾燥した容器Bに、乾燥試料を、標線Cに液面が一致するように入れ、図に示すように温度計F、補助温度計H及びかき混ぜ棒を挿入し、コルク栓で固定し、さらに、予想する凝固点より5~10℃低い温度の溶液を入れた冷却浴に浸す。以下固体の場合に準ずる。
(8) 強熱減量試験法
強熱減量試験法は、試料を各条に規定する条件で強熱し、その減量を測定する方法である。この方法は、強熱することにより、その構成成分の一部又は混在物を失う無機品目について用いる。各条に、例えば、「40.0~52.0%(1g,450~550℃,3時間)」と規定するものは、本品約1gを1mgの桁まで量り、その数値を記録し、450~550℃で3時間強熱するとき、その減量が本品1gにつき、400~520mgであることを示す。
操作法
あらかじめ、白金製、石英製又は磁製のるつぼ又は皿を各条に規定する温度で恒量になるまで強熱し、放冷した後、その質量を1mgの桁まで量り、その数値を記録する。
試料は、各条に規定する量の±10%の範囲内で採取し、前記の容器に入れ、その質量を1mgの桁まで量り、その数値を記録する。これを各条に規定する条件で強熱し、放冷した後、その質量を1mgの桁まで量り、その数値を記録する。放冷は、デシケーター(シリカゲル)で行う。
(9) 強熱残分試験法
強熱残分試験法は、試料を次の操作法により強熱するとき、揮発せずに残留する物質の量を測定する方法である。この方法は、通例、有機物中に不純物として含まれる無機物の含量を知るために用いるが、場合によっては、有機物中に構成成分として含まれる無機物又は熱時揮発する無機物中に含まれる不純物の量を測定するために用いる。
各条に、例えば、「0.1%以下(1g)」と規定するものは、本品約1gを0.1mgの桁まで量り、その数値を記録し、次の操作法により強熱するとき、その残分が本品1gにつき、1mg以下であることを示す。また、乾燥した後とあるときは、乾燥減量の項の条件で乾燥した後、測定する。
操作法
あらかじめ白金製、石英製又は磁製のるつぼを450~550℃で恒量になるまで強熱し、放冷した後、その質量を0.1mgの桁まで量り、その数値を記録する。
試料は、各条に規定する量の±10%の範囲内で採取し、前記の容器に入れ、その質量を0.1mgの桁まで量り、その数値を記録する。これに硫酸少量を加えて試料を潤し、徐々に加熱して、できる限り低温でほとんど灰化し、又は揮散した後、いったん放冷し、更に硫酸少量で潤して徐々に加熱し、白煙が生じなくなった後、450~550℃で強熱して残留物を完全に灰化する。放冷した後、その質量を0.1mgの桁まで量り、その数値を記録する。放冷は、デシケーター(シリカゲル)で行う。
各条における強熱残分の規定が%以下又はmg以下で示されている場合において、上記の操作により得た値がこの値より大きいとき又は強熱残分の規定が一定の範囲をもって示されているときは、恒量になるまで強熱を行う。
(10) 屈折率測定法
物質の屈折率とは、真空中の光の速度と物質中の光の速度との比で、物質に対する光の入射角の正弦と屈折角の正弦との比に等しい。一般に、屈折率は、光の波長及び温度により変化する。
屈折率は、空気に対する値で示し、光線としてナトリウムスペクトルD線を用い、温度t℃で測定したとき、ntDで表わす。
操作法
屈折率の測定には、通例、アッベ型屈折計を用い、各条に規定する温度の±0.2℃の範囲内で行う。
(11) 原子吸光光度法
原子吸光光度法は、光が原子蒸気層を通過するとき、基底状態の原子が特有波長の光を吸収する現象を利用し、試料中の被検元素量(濃度)を測定する方法である。
装置
通例、光源部、試料原子化部、分光部、測光部及び表示記録部からなる。また、バックグラウンド補正部を備えたものもある。光源部には、中空陰極ランプ又は放電ランプ等を用いる。試料原子化部には、フレーム方式及びフレームレス方式(電気加熱方式又は冷蒸気方式)があり、フレームレス方式(冷蒸気方式)は、還元気化法及び加熱気化法に分けられる。フレーム方式は、バーナー及びガス流量調節器からなる。フレームレス方式のうち電気加熱方式は、電気加熱炉及び電源部、冷蒸気方式は、還元気化器又は加熱気化器等の水銀発生部及び吸収セルからなる。分光部には、回折格子又は干渉フィルターを用いる。測光部は、検出器及び信号処理系からなる。表示記録部には、ディスプレイ、記録装置等がある。バックグラウンド補正部は、バックグラウンドを補正するためのもので、方式には、連続スペクトル光源方式、ゼーマン方式、非共鳴近接線方式及び自己反転方式がある。
操作法
別に規定する場合を除き、次のいずれかの方法によるものとする。
① フレーム方式
別に規定する光源ランプを装填し、測光部に通電する。光源ランプを点灯し、分光器を別に規定する分析線波長に合わせた後、適当な電流値とスリット幅に設定する。次に、別に規定する支燃性ガス及び可燃性ガスを用い、これらの混合ガスに点火してガス流量及び圧力を調節し、溶媒をフレーム中に噴霧してゼロ合わせを行う。別に規定する方法で調製した試料溶液又は標準液をフレーム中に噴霧し、その吸光度を測定する。
② フレームレス方式(電気加熱方式)
別に規定する光源ランプを装填し、測光部に通電する。光源ランプを点灯し、分光器を別に規定する分析線波長に合わせた後、適当な電流値とスリット幅に設定する。次に、別に規定する方法で調製した試料溶液又は標準液の一定量を電気加熱炉に注入し、適当な流量のフローガスを流し、適当な温度、時間及び加熱モードで、乾燥、灰化及び原子化させ、その吸光度を測定する。
③ フレームレス方式(冷蒸気方式)
別に規定する光源ランプを装填し、測光部に通電する。光源ランプを点灯し、分光器を別に規定する分析線波長に合わせた後、適当な電流値とスリット幅に設定する。次に、還元気化法では、試料溶液又は標準液を密閉容器に入れ、適当な還元剤を加えて元素になるまで還元した後、気化させる。また、加熱気化法では、試料を加熱して気化させる。これらの方法により生じた原子蒸気の吸光度を測定する。
定量は、通例、次のいずれかの方法によるものとする。なお、定量に際しては、干渉及びバックグラウンドを考慮する必要がある。
① 検量線法
3種以上の濃度の異なる標準液を調製し、それぞれの標準液につき、その吸光度を測定し、得られた値から検量線を作成する。次に、測定可能な濃度範囲に調製した試料溶液の吸光度を測定した後、検量線から被検元素量(濃度)を求める。
② 標準添加法
同量の試料溶液3個以上を量り、それぞれに被検元素が段階的に含まれるように標準液を添加し、さらに、溶媒を加えて一定容量とする。それぞれの溶液につき、吸光度を測定し、横軸に添加した標準被検元素量(濃度)、縦軸に吸光度をとり、グラフにそれぞれの値をプロットする。プロットから得られた回帰線を延長し、横軸との交点と原点との距離から被検元素量(濃度)を求める。ただし、この方法は、①による検量線が原点を通る直線の場合のみに適用できる。
③ 内標準法
内標準元素の一定量に対して、標準被検元素を段階的に加えた標準液を、数種類調製する。それぞれの液につき、各元素の分析線波長で、標準被検元素による吸光度及び内標準元素による吸光度を、同一条件で測定し、標準被検元素による吸光度と内標準元素による吸光度との比を求める。横軸に標準被検元素量(濃度)、縦軸に吸光度の比をとり、検量線を作成する。次に、標準液の場合と同量の内標準元素を加えた試料溶液を調製し、検量線を作成したときと同一条件で得た被検元素による吸光度と内標準元素による吸光度との比を求め、検量線から被検元素量(濃度)を求める。
注意:試験に用いる試薬・試液は、測定の妨げとならないものを用いる。
(12) 抗菌活性試験法
抗菌活性試験法は、飼料添加物中の酵素の抗菌活性の有無を生物学的方法により測定する試験法である。この試験に使用する水、試薬、試液、計量器、容器及びディスクは、必要に応じて無菌のものを用いる。
試験用器具
ディスクは、直径10mmのものを、メンブランフィルターは、孔径0.45μmのものを使用し、ペトリ皿は、内径90mm、高さ20mmの硬質ガラス製又は合成樹脂製であって、底面が平滑で、これに適合する蓋を有するものを使用する。
培地の種類並びにその組成及びpH
別に規定する場合を除き、次の表に掲げる組成及びpHを有するものを使用する。
| 培地の組成及びpH | |||||
| 培地1,000mLの組成 | 培地番号 | 1 | 2 | 3 | |
|
ペプトン 肉エキス 食塩 ブドウ糖 膵消化カゼイン パパイン消化大豆 リン酸一水素カリウム 寒天 蒸留水 |
(g) (g) (g) (g) (g) (g) (g) (g) |
10 5 2.5 13-15 適量 |
5 2.5 17 3 2.5 適量 |
5 15 5 13-15 適量 |
|
| 滅菌後のpH | 6.5±0.1 | 7.3±0.1 | 7.3±0.1 | ||
試験菌液の調製
① Micrococcus luteus ATCC 9341及びEscherichia coli ATCC 27166にあっては、1号培地に約1週間間隔で移植を繰り返し、35~37℃で純粋培養しながら継代保存した種菌を、使用に当たって2号培地に移植した後、35~37℃で22~24時間静置培養し、よく振り混ぜて試験菌液とする。
② Bacillus subtilis ATCC 6633にあっては、1号培地に約3か月間隔で移植を繰り返し、35~37℃で純粋培養しながら継代保存した種菌を、使用に当たって、ルー瓶中の同一培地に移し、35~37℃で1週間培養して芽胞を生じさせる。この菌苔をかき取って適量の水に均等に浮遊させ、毎分3,000回転で30分間遠心分離してその上澄液を捨てたものに、適量の水を加え、振とうした後、24時間間隔で、65℃で20分間2回加熱し、毎分1,000回転で5分間遠心分離してその上層液を採取し、芽胞数を計算し適当な濃度の芽胞浮遊液を調製する。この芽胞浮遊液を水で希釈して、1×106個/mLの芽胞浮遊液を調製し、試験菌液とする。
平板培地の調製
3種類の試験菌液1.5mLずつを量り、それぞれ一度溶かし、試験菌の活力を阻害しない温度に冷却した3号培地13.5mLに加え、完全に混和した後、滅菌ペトリ皿に注入し、水平に静置して培地を凝固させる。
操作法
試料1gに水9mLを加え、よく振り混ぜ、毎分3,000回転で5分間遠心分離し、その上澄液をメンブランフィルターを用いてろ過滅菌して試料溶液とする。ディスク6枚に試料溶液0.1mLずつを充分に吸着させ、3種類の平板培地上に各2枚ずつ対角になるように置く。さらに、ディスク6枚に水0.1mLを充分に吸着させ、2枚ずつを正方形の各頂点となるように置き、冷所で2時間放置する。次に、35~37℃で22~24時間培養し、ディスク周囲の発育阻止円の有無を観察する。
判定基準
直径12mm以上の明瞭な発育阻止円を認めたとき、抗菌活性を示すものとする。
力価試験法は、飼料添加物中の抗生物質の力価を生物学的方法又は化学的方法により測定する試験法である。別に規定する場合を除き、次の方法で試験を行う。この試験に使用する水、試薬・試液及び計量器・用器は、必要に応じ無菌のものを用いる。
円筒(カップ)
外径7.9~8.1mm、内径5.9~6.1mm、高さ9.9~10.1mmのステンレス鋼製のものを用いる。なお、円筒は、試験に支障を来すものであってはならない。
培地の種類並びにその組成及びpH
別に規定する場合を除き、次の表に掲げる組成及びpHを有するものを使用する。ただし、培地の成分として単に「ペプトン」と記載してある場合は、獣肉製ペプトン又はカゼイン製ペプトンのいずれを用いても差し支えない。培地のpHの調整は、1mol/L水酸化ナトリウム試液又は1mol/L塩酸試液を用い、滅菌後のpHが、所定のものとなるようにする。滅菌は、高圧蒸気滅菌器を用いて121℃で20分間行う。ただし、Bacillus subtilis ATCC 6633の培地は、アンモニア試液、水酸化カリウム試液又は1mol/L塩酸試液を用いて調整する。なお、既製の乾燥培地は、それぞれ規定の組成により調製した培地と同一成分を有し、同等の試験菌の発育及び性能を示す場合には、これを使用することができる。
培地の組成及びpH
| 培地 1,000mL の組成 |
培地番号 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| ペプトン(g) | 10 | 10 | 5 | 6 | 6 | 10 | 6 | 10 | 5 | 3.75 | 5 | 5 | 10 | 10 | ||||||
| 獣肉製ペプトン(g) | 6 | |||||||||||||||||||
| カゼインペプトン(g) | 4 | |||||||||||||||||||
| 肉エキス(g) | 5 | 5 | 3 | 1.5 | 1.5 | 5 | 1.5 | 10 | 5 | 3 | 5 | 5 | 1.5 | |||||||
| 塩化ナトリウム(g) | 2.5 | 2.5 | 2.5 | 2.5 | 5 | 5 | 5 | 1.25 | 80 | 2.5 | 2.5 | 50 | ||||||||
| 酵母エキス(g) | 3 | 3 | 3 | 1.25 | 2.5 | 3 | 2.5 | |||||||||||||
| ブドウ糖(g) | 1 | 1 | 5 | 1 | 2.5 | 2.5 | 5 | 1 | 1 | 10 | ||||||||||
| ポリソルベート80(mL) | 10 | 0.4 | ||||||||||||||||||
| 膵消化カゼイン(g) | 17 | 17 | ||||||||||||||||||
| パパイン消化大豆(g) | 3 | 3 | ||||||||||||||||||
| パパイン消化肝臓(g) | 0.625 | |||||||||||||||||||
| リン酸二水素カリウム(g) | 0.45 | |||||||||||||||||||
| リン酸一水素カリウム(g) | 2.5 | 2.5 | 0.69 | |||||||||||||||||
| リン酸一水素ナトリウム12水塩(g) | 2 | |||||||||||||||||||
| 硫酸マグネシウム(g) | 50 | |||||||||||||||||||
| カンテン(g) | 13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 |
13~ 20 | ||
| 水 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | |
| 滅菌後のpH | 6.4~ 6.6 |
6.9~ 7.1 |
7.9~ 8.1 |
6.4~ 6.6 |
7.9~ 8.1 |
6.4~ 6.6 |
7.9~ 8.1 |
7.2~ 7.4 |
7.2~ 7.4 |
6.4~ 6.6 |
6.9~ 7.1 |
7.2~ 7.4 |
7.9~ 8.1 |
5.9~ 6.1 |
5.9~ 6.1 |
7.9~ 8.1 |
5.9~ 6.1 |
6.4~ 6.6 |
5.9~ 6.1 |
緩衝液
緩衝液は、次に掲げる組成及びpHを有するものを滅菌して使用する。
1号緩衝液(pH4.5)
リン酸二水素カリウム13.61g(13.605~13.614g)に水約750mLを加えて溶かし、必要ならば、水酸化カリウム試液を用いてpHを4.4~4.6に調整した後、更に水を加えて1,000mLとする。
2号緩衝液(pH6.0)
リン酸二水素カリウム3.5g(3.45~3.54g)及びリン酸一水素ナトリウム12水塩3.0g(2.95~3.04g)に水約750mLを加えて溶かし、必要ならば、1mol/L水酸化ナトリウム試液又はリン酸(1→15)を用いてpHを5.9~6.1に調整した後、更に水を加えて1,000mLとする。
3号緩衝液(pH6.0)
リン酸二水素カリウム7.0g(6.95~7.04g)及びリン酸一水素ナトリウム12水塩6.0g(5.95~6.04g)に水約750mLを加えて溶かし、必要ならば、1mol/L水酸化ナトリウム試液又はリン酸(1→15)を用いてpHを5.9~6.1に調整した後、更に水を加えて1,000mLとする。
4号緩衝液(pH8.0)
リン酸一水素カリウム16.73g(16.725~16.734g)及びリン酸二水素カリウム0.523g(0.5225~0.5234g)又は無水リン酸一水素ナトリウム13.2g(13.15~13.24g)及びリン酸二水素カリウム0.91g(0.905~0.914g)に水約750mLを加えて溶かし、必要ならば、リン酸を用いてpHを7.9~8.1に調整した後、更に水を加えて1,000mLとする。
5号緩衝液(pH6.0)
リン酸二水素カリウム80g(79.5~80.4g)及びリン酸一水素カリウム20g(19.5~20.4g)に水約750mLを加えて溶かし、必要ならば、水酸化カリウム溶液(1→10)を用いてpHを5.9~6.1に調整した後、更に水を加えて1,000mLとする。
6号緩衝液(pH8.0)
リン酸二水素カリウム13.3g(13.25~13.34g)及び塩化ナトリウム100g(99.5~100.4g)に水約750mLを加えて溶かしたものに、水酸化カリウム試液を92mL加え、必要ならば、水酸化カリウム試液を用いてpHを7.9~8.1に調整した後、更に水を加えて1,000mLとする。
7号緩衝液(pH7.0)
リン酸二水素カリウム6.4g(6.35~6.44g)及びリン酸一水素ナトリウム12水塩18.9g(18.85~18.94g)に水約900mLを加えて溶かし、必要ならば、水酸化カリウム試液又はリン酸を用いてpHを6.9~7.1に調整した後、更に水を加えて1,000mLとする。
8号緩衝液(pH4.0)
乳酸9.01g(9.005~9.014g)に水約900mLを加えて溶かしたものに、1mol/L水酸化ナトリウム試液50mLを加え、必要ならば、1mol/L水酸化ナトリウム試液又はリン酸を用いてpHを3.9~4.1に調整した後、更に水を加えて1,000mLとする。
9号緩衝液(pH7.5)
2号緩衝液に1mol/L水酸化ナトリウム試液を加えてpHを7.4~7.6に調整する。
標準品及び常用標準品
標準品は、常用標準の力価を定めるための標準として、常用標準品は、抗菌性物質の力価を定めるための標準として、独立行政法人農林水産消費安全技術センターが指定する特定製造番号の抗菌性物質である。
標準品及び常用標準品は、次のとおりであり、それぞれの右欄にそのものの本質等を参考として付記する。
| 標準品名 | 標準品の本質等 | 常用標準品名 | 常用標準品の本質等 |
|---|---|---|---|
| 標準アビラマイシン | アビラマイシンA(C61H88Cl2O32) | 常用標準アビラマイシン | アビラマイシン |
| 標準エンラマイシン | 一塩酸エンラマイシン 〔一塩酸エンラマイシンA:C107H138Cl2N26O31・HCl(58%)、 一塩酸エンラマイシンB:C108H140Cl2N26O31・HCl(42%)〕 |
常用標準エンラマイシン | 一塩酸エンラマイシン |
| 標準サリノマイシン | サリノマイシンナトリウム(C42H69O11Na) | 常用標準サリノマイシン | サリノマイシンナトリウム |
| 標準センデュラマイシン | センデュラマイシンナトリウム(C45H75O16Na) | 常用標準センデュラマイシン | センデュラマイシンナトリウム |
| 標準ナラシン | ナラシンA(C43H72O11) | 常用標準ナラシン | ナラシンA |
| 標準ノシヘプタイド | ノシヘプタイド(C51H43N13O12S6) | 常用標準ノシヘプタイド | ノシヘプタイド |
| 標準ビコザマイシン | ビコザマイシン(C12H18N2O7) | 常用標準ビコザマイシン | ビコザマイシン |
| 標準フラボフォスフォリポール | フラボフォスフォリポール(C65~75H124~135N6~7O40~42P) | 常用標準フラボフォスフォリポール | フラボフォスフォリポール |
| 標準モネンシン | モネンシンナトリウム(モネンシンAナトリウム:C36H61O11Na) | 常用標準モネンシン | モネンシンナトリウム |
| 標準ラサロシド | ラサロシドナトリウム(C34H53O8Na) | 常用標準ラサロシド | ラサロシドナトリウム |
各抗菌性物質の定義
① アビラマイシン
Streptomyces viridochromogenes の培養により得られるアビラマイシンA(C61H88Cl2O32)及びアビラマイシンB(C59H84Cl2O32)を主成分とするもの又はその他の方法により得られるこれと同一の物質をいう。
② エンラマイシン
Streptomyces fungicidicus の培養により得られるエンラマイシンA(C107H138Cl2N26O31)及びエンラマイシンB(C108H140Cl2N26O31)を主成分とするもの又はその他の方法により得られるこれと同一の物質をいう。
③ サリノマイシン
Streptomyces albus の培養により得られるサリノマイシン(C42H70O11)又はその他の方法により得られるこれと同一の物質をいう。
④ センデュラマイシン
Actinomadura roseorufa の培養により得られるセンデュラマイシン(C45H76O16又はその他の方法により得られるこれと同一の物質をいう。
⑤ ナラシン
Streptomyces aureofaciens の培養により得られるナラシンA(C43H72O11)を主成分とするもの又はその他の方法により得られるこれと同一の物質をいう。
⑥ ノシヘプタイド
Streptomyces actuosus の培養により得られるノシヘプタイド(C51H43O12N13S6又はその他の方法により得られるこれと同一の物質をいう。
⑦ ビコザマイシン
Streptomyces griseoflavus の培養により得られるビコザマイシン(C12H18N2O7)又はその他の方法により得られるこれと同一の物質をいう。
⑧ フラボフォスフォリポール
Streptomyces bambergiensis の培養により得られるフラボフォスフォリポール(C65~75H124~135N6~7O40~42P)又はその他の方法により得られるこれと同一の物質をいう。
⑨ モネンシン
Streptomyces cinnamonensis の培養により得られるモネンシンA(C36H62O11)を主成分とするもの又はその他の方法により得られるこれと同一の物質をいう。
⑩ ラサロシド
Streptomyces lasaliensis の培養により得られるラサロシド(C34H54O8)又はその他の方法により得られるこれと同一の物質をいう。
各抗菌性物質の力価の定義
① アビラマイシン
アビラマイシンの力価は、アビラマイシンA(C61H88Cl2O32)としての量を質量(力価)で示す。1μg(力価)は、標準アビラマイシン1μgに相当する。
② エンラマイシン
エンラマイシンの力価は、一塩酸エンラマイシン〔一塩酸エンラマイシンA(C107H138Cl2N26O31・HCl)58%、一塩酸エンラマイシンB(C108H140Cl2N26O31・HCl)42%〕としての量を質量(力価)で示す。1μg(力価)は、0.13kPa以下の減圧下で、60℃、4時間乾燥した標準エンラマイシン1μgに相当する。
③ サリノマイシン
サリノマイシンの力価は、サリノマイシンナトリウム(C42H69O11Na)としての量を質量(力価)で示す。1μg(力価)は、0.67kPa以下の減圧下で、60℃、3時間乾燥した標準サリノマイシン1μgに相当する。
④ センデュラマイシン
センデュラマイシンの力価は、センデュラマイシン(C45H76O16Na)としての量を質量(力価)で示す。1μg(力価)は、0.67kPa以下の減圧下で、100℃、3時間乾燥した標準センデュラマイシン1μgに相当する。
⑤ ナラシン
ナラシンの力価は、ナラシンA(C43H72O11)としての量を質量(力価)で示す。1μg(力価)は、標準ナラシン1μgに相当する。
⑥ ノシヘプタイド
ノシヘプタイドの力価は、ノシヘプタイド(C51H43O12N13S6としての量を質量(力価)で示す。1μg(力価)は、標準ノシヘプタイド1μgに相当する。
⑦ ビコザマイシン
ビコザマイシンの力価は、ビコザマイシン(C12H18N2O7)としての量を質量(力価)で示す。1μg(力価)は、標準ビコザマイシン1μgに相当する。
⑧ フラボフォスフォリポール
フラボフォスフォリポールの力価は、フラボフォスフォリポール(C65~75H124~135N6~7O40~42P)としての量を質量(力価)で示す。1μg(力価)は、標準フラボフォスフォリポール1μgに相当する。
⑨ モネンシン
モネンシンの力価は、モネンシンA(C36H62O11)としての量を質量(力価)で示す。1μg(力価)は、標準モネンシン1.064μgに相当する。
⑩ ラサロシド
ラサロシドの力価は、ラサロシドナトリウム(C34H53O8Naとしての量を質量(力価)で示す。1μg(力価)は、標準ラサロシド1μgに相当する。
菌液又は胞子液の調製
試験菌としてMicrococcus luteus ATCC 9341、Micrococcus luteus ATCC 10240、Escherichia coli ATCC 27166、Bordetella bronchiseptica ATCC 4617、Corynebacterium xerosis NCTC 9755、Bacillus subtilis ATCC 6633、Bacillus brevis ATCC 8185又はBacillus cereus ATCC 19637を用いるときは、別に規定する場合を除き、次の方法により菌液又は胞子液を調製する。
① Micrococcus luteus ATCC 9341、Micrococcus luteus ATCC 10240、Escherichia coli ATCC 27166又はBordetella bronchiseptica ATCC 4617の菌液の調製
18号培地に約1週間間隔で移植を繰り返し、32~37℃で培養しながら継代保存した試験菌を、使用に当たって2号培地に接種し、32~37℃で16~24時間培養し、菌液とする。あるいは、継代培養した試験菌を、18号培地約9mLを入れた試験管斜面寒天培地(内径16mm)に接種し、32~37℃で16~24時間培養した後、この試験管斜面寒天培地に生理食塩液10mLを加え、発育した菌を洗い落として他の試験管に移し、菌液とする。菌液は、5℃以下に保存し、Micrococcus luteus ATCC 9341にあっては5日以内に、その他の菌にあっては7日以内に使用する。
② Corynebacterium xerosis NCTC 9755 の菌液の調製
1号培地に35~37℃で16~24時間3回継代培養する。これを2号培地に移植し、35~37℃で3~4時間振とう培養(振幅5cm、振動数110往復/1分)し、菌液とする。菌液は、用時調製する。
③ Bacillus subtilis ATCC 6633及びBacillus brevis ATCC 8185の胞子液の調製
1号培地に約3か月間隔で移植を繰り返し、32~37℃で培養しながら継代保存した試験菌を、ルー瓶に入れた同培地に接種し、32~37℃で1週間以上培養して胞子を作らせる。
この胞子を、生理食塩液100mLに浮遊して、65℃で30分間加熱する。遠心分離を行い、胞子を採り、更に生理食塩液約50mLずつで3回遠心分離して洗った後、生理食塩液100mLに浮遊して、65℃で30分間加熱し、胞子液とする。胞子液は、5℃以下に保存し、6か月以内に使用する。
④ Bacillus cereus ATCC 19637 の胞子液の調製
1号培地に約2週間間隔で移植を繰り返し、27~29℃で培養しながら継代保存した試験菌を、ルー瓶に入れた同培地に接種し、27~29℃で1週間以上培養して胞子を作らせた後、室温(約25℃)に約1週間放置する。この胞子を生理食塩液100mLに浮遊して、65℃で30分間加熱する。遠心分離を行い、胞子を採り、更に生理食塩液50mLずつで3回遠心分離して洗った後、生理食塩液100mLに浮遊し、65℃で30分間加熱し、胞子液とする。胞子液は、5℃以下に保存し、6か月以内に使用する。
円筒寒天平板の調製
別に規定する場合を除き、内径約90mmのペトリ皿を用いる場合は、基層用培地20mLを、内径約100mmのペトリ皿を用いる場合は、同培地21mLを、また、大型皿を用いる場合は、培地の厚さが2~3mmとなるように同培地を分注し、培地を平らに行き渡らせ、水平に静置して固化させ、基層とする。一度溶かし、試験菌の活力を阻害しない温度に冷却した各条に規定する種層用培地に、菌液又は胞子液を加え、十分に混和した後、ペトリ皿においては4mLを、大型皿においてはその厚さが1.5~2.5mmとなるように固めた基層上に分注し、基層の上面に一様に広がるように操作した後、水平に静置して培地を固化させ平板とする。4個の円筒(カップ)を、平板上に(内径約90mmのペトリ皿の場合には、半径約25mm、内径約100mmのペトリ皿の場合には、半径約28mmの円周上に)、隣り合う各々が中心に対して約90°となるように置き、円筒寒天平板を作る。また、大型皿平板を用いる場合は、ペトリ皿平板に準ずる配置に円筒(カップ)を置き、4個1組でペトリ皿1枚分とする。平板上に円筒(カップ)を置く際には、円筒(カップ)を10~13mmの高さから垂直に落とす。種層用培地に添加する菌液又は胞子液の量は、円筒寒天平板を培養したとき、高濃度の常用標準希釈液による阻止円の直径が20~25mmに、低濃度の常用標準希釈液による阻止円の直径が15~20mmになるように調整する。なお、円筒寒天平板の代わりに、平板に平板器底に達する直径7.9~8.1mmの円形のせん孔を施したせん孔寒天平板を用いることができる。
なお、円筒寒天平板の代わりに、平板に平板器底に達する直径7.9~8.1mmの円形のせん孔を施したせん孔寒天平板を用いることができる。
常用標準希釈液の調製
常用標準希釈液は、常用標準品適量を量り、各条の規定に従い、調製した希釈原液を使用に当たって高低2種類の規定濃度に希釈した液である(以下、高濃度の希釈液を「SH」、低濃度の希釈液を「SL」という。)なお、常用標準品を量る場合には、別に規定する場合を除き、相対湿度50%以下の大気中で量り、化学はかりを用いる場合の秤取量は、次の表の常用標準品の秤取量の欄に掲げる量とし、同表の常用標準品の予備乾燥条件の欄に乾燥条件が記載されている場合にあっては、当該条件であらかじめ乾燥した後、規定量を量りとる。
また、希釈原液は、原則としてそれぞれ次の表の希釈原液の保存温度の欄に掲げる温度で保存して有効期間内に使用するものとし、常用標準希釈液は、用時調製する。
| 常用標準品名 | 常用標準品の秤取量 | 常用標準品の予備乾燥条件 | 希釈原液の保存温度 | 希釈原液の有効期間 |
|---|---|---|---|---|
| 常用標準アビラマイシン | 約20mg(力価)相当量以上 | 2.67~3.33kPa,60℃,3時間 | 5℃以下 | 30日 |
| 常用標準エンラマイシン | 約20mg以上 | 0.27kPa以下,60℃,3時間 | 5℃以下 | 7日 |
| 常用標準サリノマイシン | 約20mg(力価)相当量以上 | 0.67kPa以下,60℃,3時間 | 5℃以下 | 14日 |
| 常用標準センデュラマイシン | 約25mg(力価)相当量以上 | 0.67kPa以下,100℃,3時間 | 5以下 | 7日 |
| 常用標準ナラシン | 約25mg(力価)相当量以上 | - | 5℃以下 | 14日 |
| 常用標準ノシヘプタイド | 約20mg以上 | 0.67kPa以下,60℃,3時間 | 5℃以下 | 14日 |
| 常用標準ビゴザマイシン | 約20mg以上 | - | 5℃以下 | 4日 |
| 常用標準フラボフォスフォリポール | 約20mg以上 | - | 10℃以下 | 14日 |
| 常用標準モネンシン | 約20mg(力価)相当量以上 | - | 5℃以下 | 14日 |
| 常用標準ラサロシド | 約20mg以上 | - | 5℃以下 | 30日 |
試料溶液の調製
各条で規定する(以下高濃度の試料液を「UH」、低濃度の試料液を「UL」という。)。なお、調製された試料原液又は試料溶液は、調製日の翌日以降、試験の用に供してはならない。
操作法
円筒寒天平板5枚(大型皿円筒寒天平板を用いる場合は、これに準ずる配置区分)を使用する。次の図に示す各円筒寒天平板の第1の円筒にはSHを、第2の円筒にはUHを、第3の円筒にはSLを、第4の円筒にはULをそれぞれ満たし、注意してこれをふ卵器に収め、Bacillus cereus にあっては27~29℃で、その他の菌にあっては32~37℃で、16~20時間培養する。培養後、各阻止円の直径を少なくとも0.25mmの精度で測定する。必要ならば、測定した阻止円の直径を、次の表に例示する様式のカードに記入する。なお、せん孔寒天平板の各孔に注入する常用標準希釈液及び試料溶液は、各一定量ずつで満たす。
| 円筒番号 | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 円筒内容 | 高濃度常用 標準希釈液 SH |
低濃度常用 標準希釈液 SL |
高濃度試料液 UH |
低濃度試料液 UL |
| 寒天平板 | ||||
| Ⅰ Ⅱ Ⅲ Ⅳ Ⅴ |
mm |
mm |
mm |
mm |
| 計 | ΣSH | ΣSL | ΣUH | ΣUL |
力価計算
円筒内の液体の力価(P)と阻止円の直径(d)との間には、次の関係が成立している。
d=αlogP+β(α及びβは定数)
必要に応じて、この関係式を確かめ、採取した試料中の力価を次式により求める。
採取した試験品の力価=A×高濃度常用標準希釈液1mL中の力価×高濃度試料溶液の希釈倍率
ただし、
logA=(I×V)/W
I=log(SHの力価/SLの力価)
V=ΣUH+ΣUL-ΣSH-ΣSL
W=ΣUH+ΣSH-ΣUL-ΣSL
SH、SL、UH及びULの各円筒平板の阻止円直径(mm)の和をそれぞれΣSH、ΣSL、ΣUH及びΣULとする。
(14) 酵素力試験法
① キシラン糖化力試験法
キシラン糖化力試験法は、キシランにキシラナーゼが作用するときに、加水分解に伴って増加する還元力により、飼料添加物中のキシラナーゼの量を測定する方法であり、その単位は、キシラン糖化力単位で示す。
1キシラン糖化力単位は、キシラナーゼがキシランに40℃で作用するとき、反応初期の1分間に1μmolのキシロースに相当する還元力の増加をもたらす酵素量に相当する。
基質溶液の調製
キシラン4.0g(3.95~4.04g)を量り、1mol/L水酸化ナトリウム試液50mLに徐々に加えながら激しく振り混ぜて溶かし、フェノールフタレイン試液2滴を加え、1mol/L塩酸試液で中和した後、試料の最大酵素活性を示すpHに調整した0.1mol/L酢酸・酢酸ナトリウム緩衝液100mLを加え、200mLの全量フラスコに入れ、さらに、水を標線まで加えて200mLとする。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が0.1~0.2キシラン糖化力単位となるように、水又は試料の最大酵素活性を示すpHに調整した0.01mol/L酢酸・酢酸ナトリウム緩衝液を加えて溶かし、試料溶液とする。試料が完全に溶けない場合には、ときどきかき混ぜながら1時間放置した後、遠心分離し、その上澄液を試料溶液とする。基質溶液2mLを全量ピペットを用いて量り、25×200mmの試験管に入れ、40±0.2℃の水浴中に5分間放置した後、全量ピペットを用いて試料溶液1mLを加え、よく振り混ぜ、30秒以内に40±0.2℃の水浴中で正確に30分間放置する。次に、硫酸(6→100)0.5mLを加え、よく振り混ぜ、10分間放置した後、フェノールフタレイン試液1滴を加え、1mol/L水酸化ナトリウム試液で中和する。その後、水を加えて5mLとし、全量ピペットを用いてアルカリ性銅試液A5mLを加え、試験管の口をアルミホイルで軽く覆い、ときどき振り混ぜながら20分間水浴中で加熱した後、20~30℃に冷却する。放冷した後、ヨウ化カリウム溶液(1→40)2mLを加え、さらに、硫酸(6→100)1.5mLを加え、30秒以内に激しく振り混ぜる。溶液が澄明になった後、0.005mol/Lチオ硫酸ナトリウム溶液で滴定する(指示薬 デンプン試液1mL)。この場合において、滴定の終点は、溶液の青色が消えたときとし、その滴定量をAmLとする。
別に、基質溶液2mLを全量ピペットを用いて量り、25×200mmの試験管に入れ、硫酸(6→100)0.5mLを加え、よく振り混ぜた後、全量ピペットを用いて試料溶液1mLを加え、よく振り混ぜる。次に、フェノールフタレイン試液1滴を加え、以下同様の方法で操作し、その滴定量をBmLとする。0.005mol/Lチオ硫酸ナトリウム溶液の消費量の差(B-A)(mL)に対応するキシロースの量(g)を検量線から求め、Kとする。
1g中のキシラン糖化力単位=(K/(150.13×10-6×30))×(1/W)
W:試料溶液1mL中の試料の量(g)
検量線の作成
キシロースを105℃で3時間乾燥し、その0.50g(0.495~0.504g)を量り、100mLの全量フラスコに入れ、水を加えて溶かし、更に水を標線まで加えて100mLとする。この溶液1mL、2mL、3mL及び4mLを全量ピペットを用いて量り、それぞれ100mLの全量フラスコに入れ、それぞれに水を標線まで加えて100mLとする。それぞれの溶液5mLを全量ピペットを用いて量り、それぞれ25×200mmの試験管に入れ、全量ピペットを用いてアルカリ性銅試液A5mLを加え、以下試料と同様の方法で操作し、その滴定量をそれぞれS1mL、S2mL、S3mL及びS4mLとする。別に、水5mLを全量ピペットを用いて量り、25×200mmの試験管に入れ、全量ピペットを用いてアルカリ性銅試液A5mLを加え、以下試料と同様の方法で操作し、その滴定量をB′mLとする。0.005mol/Lチオ硫酸ナトリウム溶液の消費量の差(B′-S1)、(B′-S2)、(B′-S3)及び(B′-S4)を縦軸に、それぞれに対応するキシロースの量(g)を横軸にとり、検量線を作成する。
② β-グルカン糖化力試験法
β-グルカン糖化力試験法は、β-グルカンにβ-グルカナーゼが作用するときに、加水分解に伴って増加する還元力により、飼料添加物中のβ-グルカナーゼの量を測定する方法であり、その単位は、β-グルカン糖化力単位で示す。
1β-グルカン糖化力単位は、β-グルカナーゼがβ-グルカンに30℃で作用するとき、反応初期の1分間に1μmolのブドウ糖に相当する還元力の増加をもたらす酵素量に相当する。
基質溶液の調製
β-グルカン1.0g(0.95~1.04g)を量り、100mLの全量フラスコに入れ、10mLのエタノールで湿らせた後、約80mLの水を加え、沸騰させ、β-グルカンを溶解する。かき混ぜながら室温に戻し、水を全量フラスコの標線まで加えて100mLとし、ガラスろ過器(G3)でろ過する。冷蔵庫中で保存し2日以内に使用する。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が0.1~0.2β-グルカン糖化力単位となるように、試料の最大酵素活性を示すpHに調整した0.1mol/L酢酸・酢酸ナトリウム緩衝液を加えて溶かし、試料溶液とする。試料が完全に溶けない場合には、1時間かき混ぜた後、遠心分離し、その上澄液を試料溶液とする。基質溶液1mLを全量ピペットを用いて量り、16×200mmの試験管に入れ、30±0.2℃の水浴中に5分間放置した後、全量ピペットを用いて試料溶液1mLを加え、よく振り混ぜ、30秒以内に30±0.2℃の水浴中で正確に10分間放置する。次に、全量ピペットを用いてジニトロサリチル酸試液3mLを加え、よく振り混ぜ、試験管の口をアルミホイルで軽く覆い、正確に5分間煮沸した後、冷水浴中で冷却し、室温に戻す。この溶液につき、波長540nmにおける吸光度ATを測定する。
別に、基質溶液1mLを全量ピペットを用いて量り、16×200mmの試験管に入れ、全量ピペットを用いてジニトロサリチル酸試液3mLを加え、よく振り混ぜた後、全量ピペットを用いて試料溶液1mLを加え、よく振り混ぜ、30秒以内に30±0.2℃の水浴中に正確に10分間放置する。以下同様の方法で操作し、吸光度AT′を測定する。
1g中のβ-グルカン糖化力単位=((AT-AT′)/(10×0.18))×(1/W)×F
W:試料溶液1mL中の試料の量(g)
F:検量線から求めた吸光度差1に対するブドウ糖の量(mg)
検量線の作成
あらかじめブドウ糖約1gを0.01gの桁まで量り、その数値を記録し、105℃で6時間乾燥し、その減量を測定する。その乾燥物1gに相当するブドウ糖を0.01gの桁まで量り、水を加えて溶かし、100mLの全量フラスコに入れ、更に水を標線まで加えて100mLとする。この溶液1mL、2mL、3mL、4mL、5mL及び6mLを全量ピペットを用いて量り、それぞれ100mLの全量フラスコに入れ、それぞれに水を標線まで加えて100mLとする。それぞれの溶液1mL及び水1mLを全量ピペットを用いて量り、それぞれ16×200mmの試験管に入れ、全量ピペットを用いてジニトロサリチル酸試液3mLをそれぞれ加え、よく振り混ぜ、試験管の口をアルミホイルで軽く覆い、正確に5分間煮沸した後、冷水浴中で冷却し、室温に戻す。これらの溶液につき、波長540nmにおける吸光度A1、A2、A3、A4、A5及びA6を測定する。別に、水2mLを全量ピペットを用いて量り、16×200mmの試験管に入れ、全量ピペットを用いてジニトロサリチル酸試液3mLを加え、以下同様の方法で操作して吸光度A0を測定する。吸光度差(A1-A0)、(A2-A0)、(A3-A0)、(A4-A0)、(A5-A0)及び(A6-A0)を縦軸に、ブドウ糖の量(mg)を横軸にとり、検量線を作成する。
③ 脂肪消化力試験法
脂肪消化力試験法は、オリブ油にリパーゼが作用するときに、加水分解に伴って増加する脂肪酸の量により、飼料添加物中のリパーゼの量を測定する方法であり、その単位は、脂肪消化力単位で示す。
1脂肪消化力単位は、リパーゼがオリブ油に作用するとき、反応初期の1分間に1μmolの脂肪酸に相当する消化力の増加をもたらす酵素量に相当する。
基質溶液の調製
ポリビニルアルコール試液・オリブ油混液(3:1)200~300mLを量り、乳化機の500mL容器に入れ、10℃以下に冷却しながら、毎分12,000~16,000回転で10分間乳化し、冷所で1時間放置した後、油層が分離しないことを確認した後使用する。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が1.0~5.0脂肪消化力単位となるように冷水を加えて溶かし、試料溶液とする。必要ならば、ろ過又は遠心分離を行う。基質溶液5mL及び試料の最大酵素活性を示すpHに調整した0.1mol/Lリン酸塩緩衝液4mLを全量ピペットを用いて量り、よく混合し、37±0.5℃で10分間放置した後、全量ピペットを用いて試料溶液1mLを加え、30秒以内に振り混ぜ、37±0.5℃で正確に20分間放置する。次に、アセトン・エタノール混液(1:1)10mLを加え、振り混ぜ、全量ピペットを用いて0.05mol/L水酸化ナトリウム試液10mLを加え、更に全量ピペットを用いてアセトン・エタノール混液(1:1)10mLを加え、振り混ぜた後、0.05mol/L塩酸で滴定する(指示薬 フェノールフタレイン試液2~3滴)。この場合において、滴定の終点は、溶液の赤色が消えたときとし、その滴定量をAmLとする。別に、基質溶液5mL及び試料の最大酵素活性を示すpHに調整した0.1mol/Lリン酸塩緩衝液4mLを全量ピペットを用いて量り、よく混合し、37±0.5℃で30分間放置する。次に、全量ピペットを用いてアセトン・エタノール混液(1:1)10mLを加え、振り混ぜ、さらに、全量ピペットを用いて試料溶液1mLを加え、振り混ぜ、以下同様の方法で操作し、その滴定量をBmLとする。
1g中の脂肪消化力単位=50×(B-A)×(1/20)×(1 /W)
W:試料溶液1mL中の試料の量(g)
④ 繊維糖化力試験法
繊維糖化力試験法は、カルボキシメチルセルロースナトリウムにセルラーゼが作用するときに、加水分解に伴って増加する還元力により、飼料添加物中のセルラーゼの量を測定する方法であり、その単位は、繊維糖化力単位で示す。
1繊維糖化力単位は、セルラーゼがカルボキシメチルセルロースナトリウムに37℃で作用するとき、反応初期の1分間に1μmolのブドウ糖に相当する還元力の増加をもたらす酵素量に相当する。
基質溶液の調製
あらかじめカルボキシメチルセルロースナトリウム約1gを0.001gの桁まで量り、その数値を記録し、105℃で4時間乾燥し、その減量を測定する。その乾燥物0.625gに相当するカルボキシメチルセルロースナトリウムを0.001gの桁まで量り、水50mLにかき混ぜながら少量ずつ加えた後、60~70℃でときどきかき混ぜながら20分間加温して溶かし、放冷した後、100mLの全量フラスコに移し、試料の最大酵素活性を示すpHに調整した1mol/L酢酸・酢酸ナトリウム緩衝液10mLを加え、更に水を標線まで加えて100mLとする。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が、0.02~0.08繊維糖化力単位となるように水又は試料の最大酵素活性を示すpHに調整した0.1mol/L酢酸・酢酸ナトリウム緩衝液を加えて溶かし、試料溶液とする。試料が完全に溶けない場合には、ときどきかき混ぜながら1時間放置した後、遠心分離し、その上澄液を試料溶液とする。基質溶液4mLを全量ピペットを用いて量り、25mLの全量フラスコに入れ、37±0.5℃で10分間放置した後、全量ピペットを用いて試料溶液1mLを加え、30秒以内に振り混ぜ、37±0.5℃で正確に30分間放置する。次に、アルカリ性銅試液B2mLを加え、振り混ぜ、全量フラスコに栓をし、水浴中で30分間加熱し、水冷する。さらに、ヒ素モリブデン酸試液2mLを加え、よく振り混ぜ、0.5mol/L水酸化ナトリウム試液3mLを加え、振り混ぜて沈殿を溶かし、20分間放置した後、試料の最大酵素活性を示すpHに調整した1mol/L酢酸・酢酸ナトリウム緩衝液を標線まで加えて25mLとする。この溶液1mLを全量ピペットを用いて量り、試料の最大酵素活性を示すpHに調整した1mol/L酢酸・酢酸ナトリウム緩衝液9mLを加え、よく振り混ぜ、波長750nmにおける吸光度ATを測定する。別に、試料溶液1mLを全量ピペットを用いて量り、25mLの全量フラスコに入れ、アルカリ性銅試液B2mLを加え、振り混ぜ、全量ピペットを用いて基質溶液4mLを加え、振り混ぜ、全量フラスコに栓をし、水浴中で30分間加熱し、水冷する。以下同様の方法で操作し、吸光度AT′を測定する。AT及びATに対応するブドウ糖の量(mg)を検量線から求め、それぞれGT及びGT′とする。
1g中の繊維糖化力単位=((GT-GT′)/30)×(1/0.18)×(1/W)
W:試料溶液1mL中の試料の量(g)
検量線の作成
あらかじめブドウ糖約1gを0.01gの桁まで量り、その数値を記録し、105℃で6時間乾燥し、その減量を測定する。その乾燥物1gに相当するブドウ糖を0.01gの桁まで量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、更に水を標線まで加えて1,000mLとする。この溶液1mL、2mL、3mL、4mL及び5mLを全量ピペットを用いて量り、10mLの全量フラスコに入れ、それぞれに水を標線まで加えて10mLとする。それぞれの溶液1mL、基質溶液4mL及びアルカリ性銅試液B2mLを全量ピペットを用いて量り、25mLの全量フラスコに入れ、振り混ぜ、全量フラスコに栓をし、水浴中で30分間加熱する。水冷後、ヒ素モリブデン酸試液2mLを加え、よく振り混ぜ、さらに、0.5mol/L水酸化ナトリウム試液3mLを加え、振り混ぜて沈殿を溶かし、20分間放置した後、試料の最大酵素活性を示すpHに調整した1mol/L酢酸・酢酸ナトリウム緩衝液を標線まで加えてそれぞれ25mLとする。これらの溶液1mLを全量ピペットを用いて量り、試料の最大酵素活性を示すpHに調整した1mol/L酢酸・酢酸ナトリウム緩衝液9mLを加え、よく振り混ぜる。これらの溶液につき、波長750nmにおける吸光度A1、A2、A3、A4及びA5を測定する。別に、水1mL、基質溶液4mL及びアルカリ性銅試液B2mLを全量ピペットを用いて量り、以下同様の方法で操作し、吸光度A0を測定する。吸光度差(A1-A0)、(A2-A0)、(A3-A0)、(A4-A0)及び(A5-A0)を縦軸に、ブドウ糖の量(mg)を横軸にとり、検量線を作成する。
⑤ 繊維崩壊力試験法
繊維崩壊力試験法は、ろ紙にセルラーゼが作用するときに、ろ紙が崩壊する時間により、飼料添加物中のセルラーゼの量を測定する方法であり、その単位は、繊維崩壊力単位で示す。
1,000繊維崩壊力単位は、セルラーゼがろ紙に37℃で作用するとき、1分間に1cm×1cmの大きさの酵素定量用ろ紙2枚を完全に崩壊する酵素量に相当する。
基質の調製
酵素定量用ろ紙(紙厚0.29~0.31mm、質量125~135g/m2、α繊維含量98.5%以上、灰分量0.05%以下、ろ水時間50~90秒/100mL、破裂強度1.2~1.8kg/cm2、吸水高度8~9cm/10分、透気度30~40秒/cm2/100mLのものをいう。)を光源を通して観察し、すきむらがなく、厚さが均一で異物のない個所を1cm×1cmの大きさに切る。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が2.8~4.0繊維崩壊力単位となるように、試料の最大酵素活性を示すpHに調整した1mol/L酢酸・酢酸ナトリウム緩衝液を加えて溶かし、試料溶液とする。試料が完全に溶けない場合には、ときどきかき混ぜながら1時間放置した後、遠心分離し、その上澄液を試料溶液とする。試料溶液5mLずつを全量ピペットを用いて量り、5本のL字型試験管に入れ、37±0.5℃で5分間放置した後、それぞれに基質を2枚ずつ入れ、毎分65回転、振幅60mm及び温度37±0.5℃で30秒以内に振とうする。適時、ろ紙の崩壊状態を観察し、ろ紙が完全に崩壊して微細な繊維となるまでの時間(分)を測定する。
1g中の繊維崩壊力単位=(1/(T×W)×1,000
T:ろ紙が完全に崩壊するまでの時間(最短と最長のものを除く。)の平均時間(分)
W:試料溶液5mL中の試料の量(g)
| L字型試験管 |
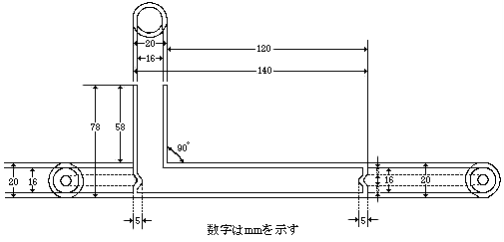 |
⑥ たん白消化力試験法
たん白消化力試験法は、カゼインにプロテアーゼが作用するときに、加水分解に伴って増加する酸可溶性分解産物の量により、飼料添加物中のプロテアーゼの量を測定する方法であり、その単位は、たん白消化力単位で示す。
1たん白消化力単位は、プロテアーゼが乳製カゼインに37℃で作用するとき、反応初期の1分間に1μgのチロシンに相当する非たん白性のフォリン試液呈色物質の増加をもたらす酵素量に相当する。
(ⅰ) 第1法
基質溶液の調製
あらかじめ乳製カゼイン約1gを0.01gの桁まで量り、その数値を記録し、105℃で2時間乾燥し、その減量を測定する。その乾燥物1.20gに相当する乳製カゼインを0.01gの桁まで量り、0.05mol/Lリン酸一水素ナトリウム試液160mLを加え、水浴中で加温して溶かす。この溶液を流水で冷却した後、1mol/L塩酸試液又は1mol/L水酸化ナトリウム試液を加えて試料の最大酵素活性を示すpHに調整した後、200mLの全量フラスコに入れ、水を標線まで加えて200mLとする。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が10~30たん白消化力単位となるように、水又は0.02mol/Lリン酸塩緩衝液を加えて溶かし、試料溶液とする。必要ならば、ろ過又は遠心分離を行う。基質溶液5mLを全量ピペットを用いて量り、37±0.5℃で10分間放置した後、全量ピペットを用いて試料溶液1mLを加え、30秒以内に振り混ぜ、37±0.5℃で正確に10分間放置する。次に、トリクロル酢酸試液B5mLを加え、振り混ぜ、37±0.5℃で30分間放置する。さらに、この溶液をろ過して沈殿物を完全に除去した後、ろ液2mLを全量ピペットを用いて量り、0.55mol/L炭酸ナトリウム試液5mL及びフォリン試液(1→3)1mLを加え、よく振り混ぜ、37±0.5℃で30分間放置した後、この溶液につき、波長660nmにおける吸光度ATを測定する。別に、試料溶液1mLを全量ピペットを用いて量り、トリクロル酢酸試液B5mLを加え、振り混ぜ、基質溶液5mLを正確に加え、37±0.5℃で30分間放置する。以下同様の方法で操作し、吸光度AT′を測定する。
1g中のたん白消化力単位=(AT-AT′)×F×(11/2)×(1/10)×(1/W)
F:検量線から求めた吸光度差1に対応するチロシンの量(μg)
W:試料溶液1mL中の試料の量(g)
検量線の作成
チロシン標準品を105℃で3時間乾燥し、その0.500g(0.4995~0.5004g)を量り、0.2mol/L塩酸試液を加えて溶かし、500mLの全量フラスコに入れ、更に0.2mol/L塩酸試液を標線まで加えて500mLとする。この溶液1mL、2mL、3mL及び4mLを全量ピペットを用いて量り、それぞれ100mLの全量フラスコに入れ、それぞれに0.2mol/L塩酸試液を標線まで加えて100mLとする。それぞれの溶液2mLを全量ピペットを用いて量り、0.55mol/L炭酸ナトリウム試液5mL及びフォリン試液(1→3)1mLをそれぞれ加え、37±0.5℃で30分間放置した後、これらの溶液につき、波長660nmにおける吸光度A1、A2、A3及びA4を測定する。別に、0.2mol/L塩酸試液2mLを全量ピペットを用いて量り、以下同様の方法で操作し、吸光度A0を測定する。吸光度差(A1-A0)、(A2-A0)、(A3-A0)及び(A4-A0)を縦軸に、チロシンの量(μg)を横軸にとり、検量線を作成する。
(ⅱ) 第2法
基質溶液の調製
あらかじめ乳製カゼイン約1gを0.01gの桁まで量り、その数値を記録し、105℃で2時間乾燥し、その減量を測定する。その乾燥物1.20gに相当する乳製カゼインを0.01gの桁まで量り、1mol/L乳酸試液16mL及び水146mLを加え、水浴中で加温して溶かす。この溶液を流水で冷却した後、1mol/L塩酸試液又は1mol/L水酸化ナトリウム試液を加えて試料の最大酵素活性を示すpHに調整した後、200mLの全量フラスコに入れ、水を標線まで加えて200mLとする。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が10~30たん白消化力単位となるように水、0.1mol/L乳酸塩緩衝液、酢酸塩酸緩衝液又は0.01mol/L酢酸・酢酸ナトリウム緩衝液を加えて溶かし、試料溶液とする。必要ならば、ろ過又は遠心分離を行う。基質溶液5mLを全量ピペットを用いて量り、37±0.5℃で10分間放置した後、全量ピペットを用いて試料溶液1mLを加え、30秒以内に振り混ぜ、37±0.5℃で正確に10分間放置する。次に、トリクロル酢酸試液A5mLを加え、振り混ぜ、37±0.5℃で30分間放置する。さらに、この溶液をろ過して沈殿物を完全に除去した後、ろ液2mLを全量ピペットを用いて量り、0.55mol/L炭酸ナトリウム試液5mL及びフォリン試液(1→3)1mLを加え、よく振り混ぜ、37±0.5℃で30分間放置した後、この溶液につき、波長660nmにおける吸光度ATを測定する別に、試料溶液1mLを全量ピペットを用いて量り、トリクロル酢酸試液A5mLを加え、振り混ぜ、全量ピペットを用いて基質溶液5mLを加え、37±0.5℃で30分間放置する。以下同様の方法で操作し、吸光度AT′を測定する。
1g中のたん白消化力単位=(AT-AT′)×F×(11/2)×(1/10)×(1/W)
F:検量線から求めた吸光度差1に対応するチロシンの量(μg)
W:試料溶液1mL中の試料の量(g)
検量線の作成
(ⅰ)の検量線の作成を準用する。
(ⅲ)第3法
基質溶液の調製
(ⅰ)の基質溶液の調製を準用する。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が10~30たん白消化力単位となるように水を加えて溶かし、試料溶液とする。必要ならば、ろ過又は遠心分離を行う。基質溶液5mLを全量ピペットを用いて量り、37±0.5℃で10分間放置した後、全量ピペットを用いて試料溶液1mLを加え、30秒以内に振り混ぜ、37±0.5℃で正確に10分間放置する。次に、トリクロル酢酸試液A5mLを加え、振り混ぜ、37±0.5℃で30分間放置する。さらに、この溶液をろ過して沈殿物を完全に除去した後、ろ液2mLを全量ピペットを用いて量り、0.55mol/L炭酸ナトリウム試液5mL及びフォリン試液(1→3)1mLを加え、よく振り混ぜ、37±0.5℃で30分間放置した後、この溶液につき、波長660nmにおける吸光度ATを測定する。別に、試料溶液1mLを全量ピペットを用いて量り、トリクロル酢酸試液A5mLを加え、振り混ぜ、全量ピペットを用いて基質溶液5mLを加え、37±0.5℃で30分間放置する。以下同様の方法で操作し、吸光度AT′を測定する。
1g中のたん白消化力単位=(AT-AT′)×F×(11/2)×(1/10)×(1/W)
F:検量線から求めた吸光度差1に対応するチロシンの量(μg)
W:試料溶液1mL中の試料の量(g)
検量線の作成
(ⅰ)の検量線の作成を準用する。
⑦ でんぷん糖化力試験法
でんぷん糖化力試験法は、デンプンにアミラーゼが作用するときに、加水分解に伴って増加する還元力により、飼料添加物中のアミラーゼの量を測定する方法であり、その単位は、でんぷん糖化力単位で示す。
1でんぷん糖化力単位は、アミラーゼがバレイショデンプンに37℃で作用するとき、反応初期の1分間に1mgのブドウ糖に相当する還元力の増加をもたらす酵素量に相当する。
基質溶液の調製
あらかじめバレイショデンプン約1gを0.01gの桁まで量り、その数値を記録し、105℃で2時間乾燥し、その減量を測定する。その乾燥物1gに相当するバレイショデンプンを0.01gの桁まで量り、三角フラスコに入れ、水20mLを加え、よく振り混ぜながら徐々に2mol/L水酸化ナトリウム試液5mLを加えてのり状とし、水浴中で振り混ぜながら3分間加熱した後、水25mLを加え、放冷した後、2mol/L塩酸試液で正確に中和し、試料の最大酵素活性を示すpHに調整した1mol/L酢酸・酢酸ナトリウム緩衝液10mLを加え、100mLの全量フラスコに入れ、更に水を標線まで加えて100mLとする。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が0.4~0.8でんぷん糖化力単位となるように、水又は試料の最大酵素活性を示すpHに調整した0.1mol/L乳酸塩緩衝液を加えて溶かし、試料溶液とする。必要ならば、ろ過又は遠心分離を行う。基質溶液10mLを全量ピペットを用いて量り、直径30mmの試験管に入れ、37±0.5℃で10分間放置した後、全量ピペットを用いて試料溶液1mLを加え、30秒以内に振り混ぜ、37±0.5℃で正確に10分間放置する。次に、フェーリング試液のアルカリ性酒石酸塩液2mLを加え、30秒以内に振り混ぜ、さらに、全量ピペットを用いてフェーリング試液の銅液2mLを加え、軽く振り混ぜた後、試験管の口に漏斗をのせ、水浴中で正確に15分間加熱し、30秒以内に流水で25℃以下に冷却する。さらに、濃ヨウ化カリウム試液2mL及び硫酸(1→6)2mLを加え、遊離したヨウ素を30秒以内に0.05mol/Lチオ硫酸ナトリウム溶液で滴定する(指示薬 溶性デンプン試液1~2滴)。この場合において、滴定の終点は、溶液の青色が消えたときとし、その滴定量をAmLとする。別に、基質溶液10mLの代わりに水10mLを量り、以下同様の方法で操作し、その滴定量をBmLとする。
1g中のでんぷん糖化力単位=(B-A)×1.6×(1/10)×(1/W)
W:試料溶液1mL中の試料の量(g)
⑧ フィチン酸分解力試験法
フィチン酸分解力試験法は、フィチン酸にフィターゼが作用するときに、加水分解により生成されるリン酸イオンの量により、飼料添加物中のフィターゼの量を測定する方法であり、その単位は、フィチン酸分解力単位で示す。
1フィチン酸分解力単位は、フィターゼがフィチン酸に37℃で作用するとき、反応初期の1分間に1μmolのリン酸を遊離させる酵素量に相当する。
(ⅰ)第1法
基質溶液の調製
あらかじめフィチン酸ナトリウムをデシケーター(シリカゲル)中で24時間以上乾燥し、その0.271g(0.2705~0.2714g)を量り、試料の最大酵素活性を示すpHに調整した0.2mol/L酢酸・酢酸ナトリウム緩衝液約50mLを加えて溶かし、0.2mol/L酢酸試液を加えてpH5.5に調整した後、100mLの全量フラスコに入れ、同緩衝液を標線まで加えて100mLとする。
反応停止発色液の調製
モリブデン酸アンモニウム1.236g(1.2355~1.2364g)に水を加えて溶かし、100mLとし、モリブデン酸アンモニウム試液を調製する。モリブデン酸アンモニウム試液1容量に、2.5mol/L硫酸試液1容量及びアセトン2容量を加え、よく振り混ぜ、30秒以内に氷中で冷却する。反応停止発色液は、用時調製する。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が0.04~0.06フィチン酸分解力単位となるように、試料の最大酵素活性を示すpHに調整した0.005mol/L酢酸・酢酸ナトリウム緩衝液を加えて溶かし、必要ならばろ過し、試料溶液とする。基質溶液0.5mLを全量ピペットを用いて量り、10mLの丸底遠沈管又は試験管に入れ、37±0.5℃に加温する。この基質溶液に、別に、全量ピペットを用いて37±0.5℃に加温した試料溶液0.5mLを加え、30秒以内に振り混ぜ、37±0.5℃で正確に10分間放置する。その後、全量ピペットを用いて氷中で冷却した反応停止発色液2mLを加え、よくかき混ぜ、さらに、全量ピペットを用いて1mol/Lクエン酸試液0.1mLを加え、よくかき混ぜる。この溶液につき、5分以内に水を対照液として波長380nmにおける吸光度ODTを測定する。別に、試料溶液0.5mLを全量ピペットを用いて量り、全量ピペットを用いて氷中で冷却した反応停止発色液2mLを加え、よくかき混ぜた後、全量ピペットを用いて基質溶液0.5mLを加え、よくかき混ぜ、全量ピペットを用いて1mol/Lクエン酸試液0.1mLを加え、更によくかき混ぜる。以下同様の方法で操作して吸光度ODTBを測定する。
1g中のフィチン酸分解力単位=(ODT-ODTB)×F×2×(1/10)×(1/W)×Z
F:検量線から求めた吸光度差1に対応するリン酸イオン濃度(μmol/mL)
W:試料採取量(g)
Z:希釈倍率
検量線の作成
リン酸二水素カリウムをデシケーター(シリカゲル)中で24時間以上乾燥し、その0.680g(0.6795~0.6804g)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、更に水を標線まで加えて1,000mLとする。この溶液1mL、2mL、3mL及び4mLを全量ピペットを用いて量り、50mLの全量フラスコに入れ、それぞれに水を標線まで加えて50mLとする。それぞれの溶液1mLを全量ピペットを用いて量り、全量ピペットを用いて氷中で冷却した反応停止発色液2mLを加え、よくかき混ぜ、さらに、全量ピペットを用いて1mol/Lクエン酸試液0.1mLを加え、よくかき混ぜる。この溶液につき、水を対照液として波長380nmにおける吸光度ODS1、ODS2、ODS3及びODS4を測定する。別に、水1mLを全量ピペットを用いて量り、全量ピペットを用いて氷中で冷却した反応停止発色液2mLを加え、よくかき混ぜ、さらに、全量ピペットを用いて1mol/Lクエン酸試液0.1mLを加え、よくかき混ぜる。この溶液につき、水を対照液として波長380nmにおける吸光度ODSBを測定する。リン酸イオン濃度を縦軸に、吸光度差(ODS1-ODSB)、(ODS2-ODSB)、(ODS3-ODSB)及び(ODS4-ODSB)を横軸にとり、検量線を作成する。
(ⅱ)第2法
基質溶液の調製
フィチン酸ナトリウム0.8g(0.75~0.84g)を量り、0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)を加えて溶かし、塩酸でpHを5.5に調整した後、100mLの全量フラスコに入れ、同混液を標線まで加えて100mLとする。
反応停止発色液の調製
モリブデン酸アンモニウム100g(99.5~100.4g)を水800mLに溶かし、25%アンモニア水10mLを加えた後、水を加えて1,000mLとし、モリブデン酸アンモニウム試液を調製する。バナジン酸アンモニウム2.35g(2.345~2.354g)に50℃に温めた水400mLを加えて溶かし、硝酸(1→3)20mLを加え、更に水を加えて1,000mLとし、バナジン酸アンモニウム試液とする。モリブデン酸アンモニウム試液1容量に、バナジン酸アンモニウム試液1容量及び硝酸(1→3)2容量を加え、混合する。反応停止発色液は、用時調製する。
操作法
試料溶液は、各条で規定する方法で調製する。基質溶液0.8mLをマイクロピペットを用いて量り、2mLのプラスチック製遠沈管に入れ、37±0.5℃に加温し、5分間放置する。この基質溶液に、別に、37±0.5℃に加温し5分間放置した試料溶液0.04mL及び各条に定める希釈液0.36mLをマイクロピペットを用いて加え、37±0.5℃に加温し、30分間放置する。その後、反応停止発色液0.8mLをマイクロピペットを用いて加え、よくかき混ぜ、室温で10分間放置する。さらに、毎分14,000回転で3分間遠心分離を行い、得られた上澄液につき、水を対照液として、波長415nmにおける吸光度ODTを測定する。別に、希釈液0.4mLを全量ピペット又はマイクロピペットを用いて量り、これに反応停止発色液0.8mLを加え、かき混ぜた後、基質溶液0.8mLを加え、10分間室温で放置する。以下同様の方法で操作して吸光度ODBを測定する。
1g中のフィチン酸分解力単位=(ODT-ODB)×F×(1/30)×(1/W)×Z
F:検量線から求めた吸光度差1に対応するリン酸イオン濃度(μmol/mL)
W:試料採取量(g)
Z:希釈倍率
検量線の作成
105℃で2時間乾燥させた後、デシケーターで保存したリン酸二水素カリウム0.682g(0.6815~0.6824g)を量り、希釈液を加えて溶かし、100mLの全量フラスコに入れ、更に希釈液を標線まで加えて100mLとする。この溶液3mL、6mL、12mL及び25mLを全量ピペットを用いて量り、それぞれ50mLの全量フラスコに入れ、それぞれに希釈液を標線まで加えて50mLとする。試料溶液と同様に操作法に従い、ODS1、ODS2、ODS3及びODS4を測定する。リン酸イオン濃度を縦軸に、測定したODBとの吸光度差(ODS1-ODB)、(ODS2-ODB)、(ODS3-ODB)及び(ODS4-ODB)を横軸にとり、検量線を作成する。
(ⅲ)第3法
基質溶液の調製
あらかじめフィチン酸ナトリウムをデシケーター(シリカゲル)中で24時間以上乾燥し、その1.0g(0.95~1.04g)を量り、0.2mol/Lクエン酸・クエン酸ナトリウム緩衝液(pH5.5)(クエン酸ナトリウム58.82g(58.815~58.824g)に水を加えて1,000mLとし、また、クエン酸42.02g(42.015~42.024g)に水を加えて1,000mLとし、これら2つの溶液をpH5.5となるように混合して調製)65mLを加えて溶かし、0.2mol/Lクエン酸試液(クエン酸4.2g(4.15~4.24g)に水を加えて溶かし、100mLにして調製)でpHを5.5に調整した後、100mLの全量フラスコに入れ、同緩衝液を標線まで加えて100mLとする。
発色液の調製
硫酸60mLを水1,000mL中にかき混ぜながら徐々に加えた後、放冷し、1mol/L硫酸試液を調製する。モリブデン酸アンモニウム2.5g(2.49~2.54g)に水を加えて溶かし、100mLとし、モリブデン酸アンモニウム試液を調製する。アスコルビン酸10g(9.9~10.4g)に水を加えて溶かし、100mLとし、アスコルビン酸試液を調製する。1mol/L硫酸試液3容量にモリブデン酸アンモニウム試液1容量及びアスコルビン酸試液1容量を加え、混合する。発色液は、用事調製する。
反応停止液の調製
トリクロル酢酸150g(145~154g)に水を加えて溶かし、1,000mLとする。
操作法
試料溶液は、各条で規定する方法で調製する。試料溶液0.5mLをマイクロピペットを用いて量り、12×150mmの試験管に入れ、37±0.5℃に加温し、5分間放置する。この試料溶液に基質溶液0.5mLをマイクロピペットを用いて加え、30秒以内に混合し、37±0.5℃で正確に15分間放置する。その後、反応停止液1.0mLをマイクロピペットを用いて加え、混合し、試料反応液とする。別に試験溶液0.5mLをマイクロピペットを用いて12×150mmの試験管に入れ、37±0.5℃に加温し、5分間放置する。この試料溶液に反応停止液1.0mLをマイクロピペットを用いて加えた後、基質溶液0.5mLをマイクロピペットを用いて加え、混合し、試料対照液とする。
水1.8mLをマイクロピペットを用いて量り、別の12×150mmの試験管に入れ、試料反応液0.2mLをマイクロピペットを用いて加え混合する。この溶液に発色液2.0mLをマイクロピペットを用いて加え混合し、50±0.5℃で15分間放置した後、室温まで放冷する。また、水2.0mLをマイクロピペットを用いて12×150mmの試験管に入れ、発色液2.0mLをマイクロピペットを用いて加え、混合し、同様に操作したものを対照液として、波長820nmにおける吸光度ODTを測定する。別に、試料対照液について、試料反応液と同様に操作し、吸光度ODBを測定する。
1g又はmL中のフィチン酸分解力単位=(ODT-ODB)×F×40×1/15×1/W×Z
F:検量線から求めた吸光度差1に対応するリン酸イオン濃度(μmol/mL)
W:試料採取量(g又はmL)
Z:希釈倍率
検量線の作成
105℃で2時間乾燥させた後、デシケーターで保存したリン酸二水素カリウム0.612g(0.6115~0.6124g)を量り、水を加えて溶かし、500mLの全量フラスコに入れ、更に水を標線まで加えて500mLとする。この溶液2mLを全量ピペットを用いて量り、200mLの全量フラスコに入れ、水を標線まで加えて200mLとし、標準液S1とする。標準液S1を順次水で正確に2倍、4倍、8倍及び16倍に希釈し、それぞれ標準液S2、S3、S4及びS5とする。各標準液2.0mLをマイクロピペットを用いて12×150mmの試験管に入れ、発色液2.0mLをマイクロピペットを用いて加え30秒以内に混合し、50±0.5℃で15分間放置した後、室温まで放冷する。試料溶液と同様に操作法に従い、ODS1、ODS2、ODS3、ODS4及びODS5を測定する。リン酸イオン濃度を縦軸に、測定したODBとの吸光度差(ODS1-ODB)、(ODS2-ODB)、(ODS3-ODB)、(ODS4-ODB)及び(ODS5-ODB)を横軸にとり、検量線を作成する。
(Ⅳ)第4法
基質溶液の調製
あらかじめフィチン酸ナトリウムをデシケーター(シリカゲル)中で24時間以上乾燥し、その2.0g(1.95~2.04g)を量り、0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)200mLを加えて溶かし、6mol/L塩酸でpHを5.5に調整した後、250mLの全量フラスコに入れ、同混液を標線まで加えて250mLとする。
反応停止発色液の調製
モリブデン酸アンモニウム100.0g(99.95~100.04g)に水を加えて溶かし、25%アンモニウム水10mLを加えた後、水を加えて1,000mLとし、モリブデン酸アンモニウム試液を調整する。バナジン酸アンモニウム2.35g(2.345~2.354g)に50~60℃に温めた水400mLを加えて溶かし、硝酸(1→3)20mLを加え、更に水を加えて1,000mLとし、バナジン酸アンモニウム試液とする。
モリブデン酸アンモニウム試液1容量に、バナジン酸アンモニウム試液1容量及び硝酸(1→3)2容量を加え、混合する。反応停止発色液は用時調整する。
モリブデン酸アンモニウム試液1容量に、バナジン酸アンモニウム試液1容量及び硝酸(1→3)2容量を加え、混合する。反応停止発色液は用時調整する。
操作法
試料溶液は、各条で規定する方法で調整する。試料溶液0.1mLをマイクロピペットを用いて量り、12×150mmの試験管に入れ、ポリソルベート20添加(0.01%)0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)0.3mLをマイクロピペットを用いて加え、混合し、37±0.5℃に加温し、5分間放置する。この試料溶液に、あらかじめ37±0.5℃に加温した基質溶液0.8mLをマイクロピペットを用いて加え、混合し、37±0.5℃で正確に30分間放置する。その後、反応停止発色液0.8mLをマイクロピペットを用いて加え、混合し、試験反応液とする。別に試料溶液0.1mLをマイクロピペットを用いて量り、12×150mmの試験管に入れ、ポリソルベート20添加(0.01%)0.25mol/L酢酸ナトリウム塩酸混液(pH 5.5)0.3mLをマイクロピペットを用いて加え、混合し、37±0.5℃に加温し、5分間放置する。この試料溶液に反応停止発色液0.8mLをマイクロピペットを用いて加えた後、あらかじめ37±0.5℃に加温した基質溶液0.8mLをマイクロピペットを用いて加え、混合し、試験対照溶液とする。
試験反応溶液及び試験対照溶液を室温で10分間放置した後、11,000×gで3分間遠心分離を行い、得られた上澄液につき、水を対照液として波長415nmにおける吸光度ODT及びODTBを測定する。
試験反応溶液及び試験対照溶液を室温で10分間放置した後、11,000×gで3分間遠心分離を行い、得られた上澄液につき、水を対照液として波長415nmにおける吸光度ODT及びODTBを測定する。
1g中のフィチン酸分解力単位=(ODT-ODTB)×F×1/30×1/W×Z
ODT:試験反応溶液の平均吸光度
ODTB:試験対照溶液の平均吸光度
Z:希釈倍率
F:検量線から求めた吸光度差1に対応するリン酸イオン濃度(μmol/mL)
W:試料採取量(g)
検量線の作成
リン酸二水素カリウム約10gを105℃で2時間乾燥させた後、デシケーターで保存し、その0.682g(0.6815~0.6824g)を量り、ポリソルベート20添加(0.01%)0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)を加えて溶かし、100mLの全量フラスコに入れ、さらに同混液を標線まで加えて100mLとしたものを標準原液とする。
標準原液を順次水で正確に2倍、4倍、8倍及び16倍に希釈し、標準液とする。各標準液0.04mLをマイクロピペットを用いて量り、12×150mmの試験管に入れ、ポリソルベート20添加(0.01%)0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)0.36mLをマイクロピペットを用いて加え混合した後、基質溶液0.8mL及び反応停止発色液0.8mLをマイクロピペットを用いて加え混合したものを、リン酸標準反応溶液とする。リン酸標準反応溶液を室温で10分間放置した後、11,000×gで3分間遠心分離を行い、得られた上澄液につき、水を対照液として波長415nmにおける吸光度ODS1、ODS2、ODS3及びODS4を測定する。別にポリソルベート20添加(0.01%)0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)0.4mLをマイクロピペットを用いて量り、12×150mmの試験管に入れ、基質溶液0.8mL及び反応停止発色液0.8mLをマイクロピペットを用いて加えて混合したものを、リン酸標準対照溶液とする。リン酸標準対照溶液を室温で10分間放置した後、11,000×gで3分間遠心分離を行い、得られた上澄液につき、水を対照液として波長415nmにおける吸光度ODBを測定する。リン酸イオン濃度を縦軸に、測定したリン酸標準反応溶液とリン酸標準対照溶液の吸光度差(ODS1-ODB)、(ODS2-ODB)、(ODS3-ODB)及び(ODS4-ODB)を横軸にとり、検量線を作成する。
標準原液を順次水で正確に2倍、4倍、8倍及び16倍に希釈し、標準液とする。各標準液0.04mLをマイクロピペットを用いて量り、12×150mmの試験管に入れ、ポリソルベート20添加(0.01%)0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)0.36mLをマイクロピペットを用いて加え混合した後、基質溶液0.8mL及び反応停止発色液0.8mLをマイクロピペットを用いて加え混合したものを、リン酸標準反応溶液とする。リン酸標準反応溶液を室温で10分間放置した後、11,000×gで3分間遠心分離を行い、得られた上澄液につき、水を対照液として波長415nmにおける吸光度ODS1、ODS2、ODS3及びODS4を測定する。別にポリソルベート20添加(0.01%)0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)0.4mLをマイクロピペットを用いて量り、12×150mmの試験管に入れ、基質溶液0.8mL及び反応停止発色液0.8mLをマイクロピペットを用いて加えて混合したものを、リン酸標準対照溶液とする。リン酸標準対照溶液を室温で10分間放置した後、11,000×gで3分間遠心分離を行い、得られた上澄液につき、水を対照液として波長415nmにおける吸光度ODBを測定する。リン酸イオン濃度を縦軸に、測定したリン酸標準反応溶液とリン酸標準対照溶液の吸光度差(ODS1-ODB)、(ODS2-ODB)、(ODS3-ODB)及び(ODS4-ODB)を横軸にとり、検量線を作成する。
(ⅱ)第5法
基質溶液の調製
フィチフィチン酸ナトリウム0.98g(0.975~0.984g)を量り、0.25mol/L酢酸ナトリウム緩衝液(pH5.5)約80mLを加えて溶かし、酢酸でpHを5.5に調整した後、100mLの全量フラスコに入れ、同緩衝液を標線まで加えて100mLとする。反応停止発色液の調製
モリブデン酸アンモニウム20g(19.5~20.4g)を水180mLに溶かし、アンモニア水2mLを加えた後、水を加えて200mLとし、モリブデン酸アンモニウム試液を調製する。バナジン酸アンモニウム0.47g(0.465~0.474g)に水160mLを加え、水浴中で撹拌しながら約50℃に加温して溶かした後、硝酸(175→500)4mLを加え、室温まで冷却した後、水を加えて200mLとし、バナジン酸アンモニウム試液を調製する。
モリブデン酸アンモニウム試液25mLに、バナジン酸アンモニウム試液25mLを加え、よく振り混ぜ、硝酸(175→500)16.5mLを加えた後、水を加えて100mLとする。反応停止発色液は、用時調製する。
モリブデン酸アンモニウム20g(19.5~20.4g)を水180mLに溶かし、アンモニア水2mLを加えた後、水を加えて200mLとし、モリブデン酸アンモニウム試液を調製する。バナジン酸アンモニウム0.47g(0.465~0.474g)に水160mLを加え、水浴中で撹拌しながら約50℃に加温して溶かした後、硝酸(175→500)4mLを加え、室温まで冷却した後、水を加えて200mLとし、バナジン酸アンモニウム試液を調製する。
モリブデン酸アンモニウム試液25mLに、バナジン酸アンモニウム試液25mLを加え、よく振り混ぜ、硝酸(175→500)16.5mLを加えた後、水を加えて100mLとする。反応停止発色液は、用時調製する。
操作法
試料溶液は、各条で規定する方法で調製する。基質溶液2mLを全量ピペットを用いて量り、15mLプラスチック製遠心沈殿管に入れ、37±0.5℃に加温し、5~10分間放置する。この基質溶液に、別に、37±0.5℃に加温し、5~10分間放置した試料溶液1mLを全量ピペットを用いて加え、37±0.5℃で正確に60分間放置する。その後、反応停止発色液2mLを全量ピペットを用いて加え、よくかき混ぜ、室温で20分間放置し、更に2,500×gで10分間遠心分離を行い、得られた上澄液を試験反応溶液とする。この溶液につき、水を対照液として波長415nmにおける吸光度ODTを測定する。別に、基質溶液2mLを全量ピペットを用いて量り 、15mLプラスチック製遠心沈殿管に入れ、これに反応停止発色液2mLを全量ピペットを用いて加え、かき混ぜた後、試料溶液1mLを全量ピペットを用いて加え、よくかき混ぜ、室温で20分間放置し、更に2,500×gで10分間遠心分離を行い、得られた上澄液を試験対照溶液とする。この溶液につき、水を対照液として波長415nmにおける吸光度ODBを測定する。
。を対照液として波長415nmにおける吸光度ODS1、ODS2、ODS3及びODS4を測定する。別にポリソルベート20添加(0.01%)0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)0.4mLをマイクロピペットを用いて量り、12×150mmの試験管に入れ、基質溶液0.8mL及び反応停止発色液0.8mLをマイクロピペットを用いて加えて混合したものを、リン酸標準対照溶液とする。リン酸標準対照溶液を室温で10分間放置した後、11,000×gで3分間遠心分離を行い、得られた上澄液につき、水を対照液として波長415nmにおける吸光度ODBを測定する。
。を対照液として波長415nmにおける吸光度ODS1、ODS2、ODS3及びODS4を測定する。別にポリソルベート20添加(0.01%)0.25mol/L酢酸ナトリウム塩酸混液(pH5.5)0.4mLをマイクロピペットを用いて量り、12×150mmの試験管に入れ、基質溶液0.8mL及び反応停止発色液0.8mLをマイクロピペットを用いて加えて混合したものを、リン酸標準対照溶液とする。リン酸標準対照溶液を室温で10分間放置した後、11,000×gで3分間遠心分離を行い、得られた上澄液につき、水を対照液として波長415nmにおける吸光度ODBを測定する。
1g中のフィチン酸分解力単位=(ODT-ODTB)×F×1/60×1/W×Z
ODT:試験反応溶液の平均吸光度
ODTB:試験対照溶液の平均吸光度
F:検量線から求めた吸光度差1に対応するリン酸イオン濃度(μmol/mL)
V:試料溶液の定容量(mL)
W:試料採取量(g)
Z:希釈倍率
検量線の作成
105℃で2時間乾燥させた後、デシケーターで保存したリン酸二水素カリウム0.098g(0.0975~0.0984g)を量り、0.25mol/L酢酸ナトリウム緩衝液(pH5.5)を加えて溶かし、100mLの全量フラスコに入れ、更に0.25mol/L酢酸ナトリウム緩衝液(pH5.5)を標線まで加えて100mLとした後、pH5.5であることを確認し、7.20μmol/mL標準原液を調製する。
標準原液の一定量をそれぞれ試験管に入れ、0.25mol/L酢酸ナトリウム緩衝液(pH5.5)で正確に1.25倍、2.5倍、5倍及び10倍に希釈し、標準液S1、S2、S3及びS4を調製する。
各標準液1mLを全量ピペットを用いて量り、15mLプラスチック製遠心沈殿管に入れ、基質溶液2mL及び反応停止発色液2mLを全量ピペットを用いて加え、よくかき混ぜ、室温で20分間放置し、更に2,500×gで10分間遠心分離を行い、得られた上澄液をリン酸標準反応溶液とする。試験反応液と同様に操作法に従い、ODS1、ODS2、ODS3及びODS4を測定する。別に0.25mol/L酢酸ナトリウム緩衝液(pH5.5)1mLを全量ピペットを用いて量り、15mLプラスチック製遠心沈殿管に入れ、基質溶液2mL及び反応停止発色液2mLを全量ピペットを用いて加え、よくかき混ぜ、室温で20分間放置し、更に2,500×gで10分間遠心分離を行い、得られた上澄液をリン酸標準対照溶液とする。試験反応液と同様に操作法に従い、ODSBを測定する。リン酸イオン濃度を縦軸に、測定したODSBとの吸光度差(ODS1-ODSB)、(ODS2-ODSB)、(ODS3-ODSB)及び(ODS4-ODSB)を横軸にとり、検量線を作成する。
標準原液の一定量をそれぞれ試験管に入れ、0.25mol/L酢酸ナトリウム緩衝液(pH5.5)で正確に1.25倍、2.5倍、5倍及び10倍に希釈し、標準液S1、S2、S3及びS4を調製する。
各標準液1mLを全量ピペットを用いて量り、15mLプラスチック製遠心沈殿管に入れ、基質溶液2mL及び反応停止発色液2mLを全量ピペットを用いて加え、よくかき混ぜ、室温で20分間放置し、更に2,500×gで10分間遠心分離を行い、得られた上澄液をリン酸標準反応溶液とする。試験反応液と同様に操作法に従い、ODS1、ODS2、ODS3及びODS4を測定する。別に0.25mol/L酢酸ナトリウム緩衝液(pH5.5)1mLを全量ピペットを用いて量り、15mLプラスチック製遠心沈殿管に入れ、基質溶液2mL及び反応停止発色液2mLを全量ピペットを用いて加え、よくかき混ぜ、室温で20分間放置し、更に2,500×gで10分間遠心分離を行い、得られた上澄液をリン酸標準対照溶液とする。試験反応液と同様に操作法に従い、ODSBを測定する。リン酸イオン濃度を縦軸に、測定したODSBとの吸光度差(ODS1-ODSB)、(ODS2-ODSB)、(ODS3-ODSB)及び(ODS4-ODSB)を横軸にとり、検量線を作成する。
⑨ ペクチン液化力試験法
ペクチン液化力試験法は、ペクチンにペクチナーゼが作用するときに、ペクチンの全体的低分子化に伴って低下するペクチンの粘度により、飼料添加物中のペクチンの量を測定する方法であり、その単位は、ペクチン液化力単位で示す。
1ペクチン液化力単位は、ペクチナーゼが1%のペクチン溶液に37℃で10分間作用するとき、その粘度を半減させる酵素量に相当する。
装置
測定装置として図に示す粘度計を用いる。
基質溶液の調製
あらかじめペクチン約1gを0.01gの桁まで量り、その数値を記録し、105℃で2時間乾燥し、その減量を測定する。その乾燥物2.0gに相当するペクチンを0.01gの桁まで量り、水50mLを加え、加温して溶かし、100mLの全量フラスコに入れ、放冷した後、水を標線まで加えて100mLとする。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度がそれぞれ約0.5ペクチン液化力単位、約1.0ペクチン液化力単位及び約2.0ペクチン液化力単位となるように、試料の最大酵素活性を示すpHに調整した0.2mol/L酢酸・酢酸ナトリウム緩衝液を加えて溶かし、試料溶液A、B及びCとする。
基質溶液5mL及び試料の最大酵素活性を示すpHに調整した0.2mol/L酢酸・酢酸ナトリウム緩衝液4mLを、粘度計の管Dから球Aに入れ、37±0.5℃の水浴中に10分間放置した後、試料溶液A1mLを加え、30秒以内によく振り混ぜ、37±0.5℃の水浴中に10分間放置する。粘度計の管Eから静かに吸引して液面を球Cの中心部まで引き上げた後、吸引を止め、30秒以内に重力により流下させ、液面が球Bの上の標線から下の標線まで流下するのに要する時間ta1を測定する。別に、水5mL及び試料の最大酵素活性を示すpHに調整した0.2mol/L酢酸・酢酸ナトリウム緩衝液4mLを、粘度計の管Dから球Aに入れ、37±0.5℃の水浴中に10分間放置した後、試料溶液A1mLを加え、30秒以内によく振り混ぜ、37±0.5℃の水浴中に正確に10分間放置する。以下同様の方法で粘度計を操作し、流下時間ta2を測定し、TA(ta1-ta2)を求める。次に、別に、試料溶液B及びCにつき、試料溶液Aと同様の方法で操作し、TB(tb1-tb2)及びTC(tc1-tc2)を求める。
さらに、別に、基質溶液5mL及び試料の最大酵素活性を示すpHに調整した0.2mol/L酢酸・酢酸ナトリウム緩衝液5mLを、粘度計の管Dから球Aに入れ、30秒以内によく振り混ぜ、37±0.5℃の水浴中に10分間放置する。以下同様の方法で粘度計を操作し、流下時間t01を測定する。別に、水5mL及び試料の最大酵素活性を示すpHに調整した0.2mol/L酢酸・酢酸ナトリウム緩衝液5mLを、粘度計の管Dから球Aに入れ、30秒以内によく振り混ぜ、37±0.5℃の水浴中に10分間放置する。以下同様の方法で粘度計を操作し、流下時間t02を測定し、T0(t01-t02)を求める。
片対数グラフの縦軸にTA、TB及びTCを、横軸(対数尺)に試料溶液A、B及びCそれぞれの1mL中に含まれる試料の量(g)をとり、T0/2に対応する試料の量W(g)を求める。
1g中のペクチン液化力単位=1/W
⑩ ペクチン糖化力試験法
ペクチン糖化力試験法は、ペクチンにペクチナーゼが作用するときに、加水分解に伴って増加する還元力により、飼料添加物中のペクチナーゼの量を測定する方法であり、その単位は、ペクチン糖化力単位で示す。
1ペクチン糖化力単位は、ペクチナーゼがペクチンに40℃で作用するとき、反応初期の1時間に1μmolのガラクツロン酸の還元力に相当する還元力の増加をもたらす酵素量に相当する。
基質溶液の調製
ペクチン0.65g(0.645~0.654g)を量り、試料の最大酵素活性を示すpHに調整したクエン酸塩緩衝液に徐々に加えながら激しく振り混ぜて溶かし、100mLの全量フラスコに入れ、更に同緩衝液を標線まで加えて100mLとする。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が25~35ペクチン糖化力単位となるように、試料の最大酵素活性を示すpHに調整したクエン酸塩緩衝液を加えて溶かし、試料溶液とする。試料が完全に溶けない場合には、ときどきかき混ぜながら1時間放置した後、遠心分離し、その上澄液を試料溶液とする。基質溶液10mLを全量ピペットを用いて量り、100mLのヨウ素フラスコに入れ、40±0.2℃の水浴中に5分間放置した後、全量ピペットを用いて試料溶液1mLを加え、よく振り混ぜ、30秒以内に40±0.2℃の水浴中で正確に1時間放置する。次に、炭酸ナトリウム試液2.5mLを加え、よく振り混ぜ、さらに、全量ピペットを用いて0.05mol/Lヨウ素液5mLを加え、よく振り混ぜ、40分間暗所に放置した後、硫酸(12→100)5mLを加え、よく振り混ぜ、30秒以内に0.01mol/Lチオ硫酸ナトリウム液で滴定する(指示薬 デンプン試液1mL)。この場合において、滴定の終点は、溶液の青色が消えたときとし、その滴定量をAmLとする。別に、基質溶液10mLを全量ピペットを用いて量り、100mLのヨウ素フラスコに入れ、炭酸ナトリウム試液2.5mLを加え、よく振り混ぜた後、全量ピペットを用いて試料溶液1mLを加え、よく振り混ぜ、さらに、全量ピペットを用いて0.05mol/Lヨウ素液5mLを加え、よく振り混ぜ、以下同様に操作し、その滴定量をBmLとする。
1g中のペクチン糖化力単位=〔(B-A)×2-3〕×0.01 × 513 ×(1/W)
W:試料溶液1mL中の試料の量(g)
⑪ ペプチドグリカン分解力試験法
ペプチドグリカン分解力試験法は、蛍光標識ペプチドグリカンにムラミダーゼが作用するときに、加水分解に伴って増加する蛍光強度により、飼料添加物中のムラミダーゼの量を測定する方法であり、その単位は、ペプチドグリカン分解力単位で示す。
1ペプチドグリカン分解力単位は、ムラミダーゼがフルオレセイン標識ペプチドグリカンにpH6.0、30℃で作用するとき、1分間に0.06nmolのフルオレセインイソチオシアナート(アイソマーI)に相当する蛍光強度を増加させる酵素量に相当する。
希釈液
リン酸一水素ナトリウム・二水和物22.5g(22.45~22.54g)及びクエン酸7.74g(7.735~7.744g)を量り、1Lの全量フラスコに入れ、800mLの水を加え、溶解するまで攪拌した後、オクチルフェノールエトキシレート試液1mLを加え、0.05mol/L水酸化ナトリウム試液又は0.1mol/L塩酸試液を用いてpHを5.9~6.1に調整する。さらに水を標線まで加える。
基質溶液の調製
0.5mg/mLフルオレセイン標識ペプチドグリカン試液100μLに1,900μLの希釈液を加え混合する。用時調製する。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が0.01~0.03ペプチドグリカン分解力単位となるように希釈液を加え、45~90分間かき混ぜて得られた液を試料溶液とする。
標準液A~G及び試料溶液を50μLずつマイクロプレート(黒色)に分注し、使用しない隣接ウェルには希釈液を100μL分注する。標準液A~G及び試料溶液の入ったウェルに基質溶液を速やかに50μLずつ分注し、直ちに励起波長485nm、蛍光波長528nm及び温度30℃に設定した蛍光マイクロプレートリーダーを用いて、測定間隔1分で30分間測定する。
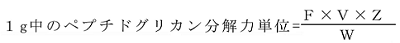
F:検量線から求めたペプチドグリカン分解力単位
V:調製した試料溶液量
Z:試料溶液の希釈倍率
W:試料採取量(g)
検量線の作成
70,000ペプチドグリカン分解力単位に相当するムラミダーゼを量り、適量の希釈液を加え、45~90分間よくかき混ぜて溶かし、100mLの全量フラスコに入れ、さらに希釈液を標線まで加えて100mLとする。この液50μLを100mLの全量フラスコに入れ、希釈液を標線まで加えて100mLとし、標準原液とする。この液を下表に従い、希釈液にて希釈し、標準液A~Gとする。
| 標準液 | 希釈倍率 | 標準原液量 (μL) |
希釈液量 (μL) |
ペプチドグリカン分解力単位/mL |
|---|---|---|---|---|
| A | 40倍 | 30 | 1,170 | 0.0088 |
| B | 30倍 | 40 | 1,160 | 0.012 |
| C | 24倍 | 50 | 1,150 | 0.015 |
| D | 20倍 | 60 | 1,140 | 0.018 |
| E | 15倍 | 80 | 1,120 | 0.023 |
| F | 12倍 | 100 | 1,100 | 0.029 |
| G | 10倍 | 120 | 1,080 | 0.035 |
標準液A~Gの0~30分の測定値から1分間当たりのそれぞれの蛍光強度増加量(傾き)を算出する。算出した傾きを縦軸に、各標準液の1mL中のペプチドグリカン分解力単位を横軸にとり、検量線を作成する。
⑫ ラクターゼ試験法
ラクターゼ試験法は、o-ニトロフェニル-β-D-ガラクトピラノシドにラクターゼが作用するときに、加水分解により生成されるo-ニトロフェノールの量により、飼料添加物中のラクターゼの量を測定する方法であり、その単位は、ラクターゼ単位で示す。
1ラクターゼ単位は、ラクターゼがo-ニトロフェニル-β-D-ガラクトピラノシドに30℃で作用するとき、反応初期の1分間に1μmolのo-ニトロフェニル-β-D-ガラクトピラノシドが加水分解する酵素量に相当する。
基質溶液の調製
o-ニトロフェニル-β-D-ガラクトピラノシド0.172g(0.1715~0.1724g)を量り、試料の最大酵素活性を示すpHに調整したマッキルベイン緩衝液に溶かし、100mLの全量フラスコに入れ、同緩衝液を標線まで加えて100mLとする。
操作法
試験を行うために必要な量の試料を有効数字3桁まで量り、その数値を記録し、1mL当たりの濃度が0.05~0.08ラクターゼ単位となるように水を加えて溶かし、試料溶液とする。試料が完全に溶けない場合には、ときどきかき混ぜながら1時間放置した後、遠心分離し、その上澄液を試料溶液とする。基質溶液3.5mLを全量ピペットを用いて量り、30℃に加温し、試料溶液0.5mLを全量ピペット又はマイクロピペットを用いて加え、30℃で正確に10分間放置した後、全量ピペットを用いて炭酸ナトリウム試液1mLを加え、振り混ぜ、この溶液につき、波長420nmにおける吸光度ATを測定する。別に、基質溶液3.5mLを全量ピペットを用いて量り、30℃に加温し、全量ピペットを用いて炭酸ナトリウム試液1mLを加え、よく振り混ぜた後、さらに、試料溶液0.5mLを全量ピペット又はマイクロピペットを用いて加え、30℃で正確に10分間放置し、この溶液につき、同様に吸光度AT′を測定する。
1g中のラクターゼ単位=((AT-AT′)/0.90)×(1/10)×(2/W)
W:試料溶液1mL中の試料の量(g)
(15) 1,4-ジオキサン試験法
1,4-ジオキサン試験法は、乳化剤中に混在する1,4-ジオキサンの限度試験である。
装置
図に示すものを用いる。
A:マノメーター
B:二方コック
C:三方コック
D:濃縮管(石英ガラス製で、正確に0.9mL又はそれ以上の蒸留物を測定し、正確に2.0mLに希釈できるように目印をつける。)
E:ナス型フラスコ
F:真空トラップ管
操作法
試料約20gを0.01gの桁まで量り、その数値を記録し、ナス型フラスコEに入れる。試料が半固体又はワックス状の場合は、水浴上で加熱し、液状とした後、ナス型フラスコEに入れる。試料が結晶状の場合は、これに1.5mLの水を加え、液状、半固体又はワックス状の場合は、1.0mLの水を加える。なお、試料の水分含量が多い場合は、水の絶対量が1.0~1.5mLとなるように調整する。マグネチックスターラーを用いて水と試料をよくかき混ぜた後、フラスコEを氷浴中に浸し、約1分間冷却する。次に、フラスコEと濃縮管Dを接続する管にリボンヒーターを巻き、約10~15Vの電圧で通電する。フラスコE及び濃縮管Dを蒸留装置に取り付け、各々のジョイントに、高圧真空用シリコーングリースを塗布する。真空トラップ管Fを液体窒素を満たしたデュワー瓶に浸し、コックBを閉じ、その後、コックCを開いて真空ポンプを作動させる。フラスコEをドライアイス・メタノール浴に約10分間浸し、真空度が0.007kPa以下になったとき、コックBを20秒間開き、再び閉じる。ドライアイス・メタノール浴を除き、約1分間室温に放置した後、20~25℃の水浴中に約5分間浸す。水浴の温度を35~40℃に上げ(大部分の試料が液状になる。)、マグネチックスターラーを用いて試料を完全に溶解させた後、氷水中で2分間冷却する。次に、水浴をドライアイス・メタノール浴に取りかえて約10分間浸し、内容物を凍結させ、コックBを20秒間開き、再び閉じる。ドライアイス・メタノール浴を除いて水浴中に浸し、水浴の温度を45~50℃に上げ、試料が完全に溶解するまで攪拌する。このとき、フラスコEと濃縮管Dとの連結管に濃縮物が認められる場合は、リボンヒーターの電圧をゆっくり上げ、濃縮物が消失するまで加熱する。さらに、試料を攪拌しながら、濃縮管Dを液体窒素を満たしたデュワー瓶に破損しないように注意してゆっくり浸す。水が管中で蒸留を始め、濃縮管Dに氷ができれば、氷の面のやや下に液体窒素面を保つようにデュワー瓶を少しずつ上げていく。濃縮管Dの首まで水が凍結する又は濃縮管Dの2.0mLの目盛りまで液体窒素面を上げるときに、デュワー瓶を除き、室温で氷を溶かす。氷が溶けた後、蒸留された水の量が0.9mL以上になるまで冷却と加温を連続して繰り返す。濃縮管Dを再び2分間冷却して内容物を凍結させ、コックCを開いて真空を解除した後、真空ポンプを停止し、コックBを開く。濃縮管Dを取り外し、栓をして室温で氷を溶かす。濃縮管Dに水を加えて正確に2.0mLとした後、均一になるまで攪拌して試料溶液とする。試料溶液及び1,4-ジオキサン測定用標準溶液につき、次の条件でガスクロマトグラフ法により試験を行い、各々のピーク高さを測定する。
操作条件
検出器:水素炎イオン化検出器
カラム:内径3mm、長さ2.0mのガラス管又はステンレス管に149~177μmのガスクロマトグラフ用多孔性アクリロニトリルジビニルベンゼン重合体又はこれと同等のものを充填する。
カラム温度:140~150℃の一定温度
キャリヤーガス及び流量:ジオキサンが約40分後に現れるように窒素の流量を調節する。
1,4-ジオキサン測定用標準溶液の調製
ジオキサン1.000g(0.9995~1.0004g)を量り、水を加えて溶かし、100mLの全量フラスコに入れ、更に水を標線まで加えて100mLとする。この溶液1mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、水を標線まで加えて100mLとする。冷所に保存し、1週間以内に使用する。
(16) 重金属試験法
重金属試験法は、試料中に混在する重金属の限度試験である。この重金属とは、酸性で硫化ナトリウム試液により呈色する金属性混在物をいい、その量は、鉛(Pb)の量として表す。
各条には、重金属(Pbとして)の限度をμg/gで( )内に付記する。
操作法
試料溶液及び比較液の調製法は、別に規定する場合を除き、次の方法によるものとする。
① 第1法
各条に規定する量の試料をネスラー管に入れ、適量の水を加えて溶かし、40mLとする。これに希酢酸2mL及び水を加えて50mLとし、試料溶液とする。
比較液は、各条に規定する量の鉛標準液をネスラー管に入れ、希酢酸2mL及び水を加えて50mLとする。
② 第2法
各条に規定する量の試料を石英製又は磁製のるつぼに量り、ゆるく蓋をし、弱く加熱して炭化する。放冷した後、硝酸2mL及び硫酸5滴を加え、白煙の生じるまで注意して加熱した後、500~600℃で強熱して灰化する。放冷した後、塩酸2mLを加え、水浴上で蒸発乾固し、残留物を塩酸3滴で潤し、熱湯10mLを加え、2分間加温する。次に、フェノールフタレイン試液1滴を加え、アンモニア試液を溶液が微赤色となるまで滴加し、希酢酸2mLを加え、必要ならば、ろ過し、水10mLで洗い、ろ液及び洗液をネスラー管に入れ、水を加えて50mLとし、試料溶液とする。
比較液は、硝酸2mL、硫酸5滴及び塩酸2mLを水浴上で蒸発し、更に砂浴上で蒸発乾固し、残留物を塩酸3滴で潤し、以下試料溶液の調製法と同様に操作し、各条に規定する量の鉛標準液及び水を加えて50mLとする。
③ 第3法
各条に規定する量の試料を石英製又は磁製のるつぼに量り、初めは注意して弱く加熱し、その後、強熱して灰化する。放冷した後、王水1mLを加え、水浴上で蒸発乾固し、残留物を塩酸3滴で潤し、熱湯10mLを加え、2分間加温する。次に、フェノールフタレイン試液1滴を加え、アンモニア試液を溶液が微赤色となるまで滴加し、希酢酸2mLを加え、必要ならば、ろ過し、水10mLで洗い、ろ液及び洗液をネスラー管に入れ、水を加えて50mLとし、試料溶液とする。
比較液は、王水1mLを水浴上で蒸発乾固し、以下試料溶液の調製法と同様に操作し、各条に規定する量の鉛標準液及び水を加えて50mLとする。
試料溶液の試験は、別に規定する場合を除き、次の方法によるものとする。
試料溶液及び比較液に硫化ナトリウム試液1滴ずつを加え、混和し、5分間放置した後、両管を白色の背景を用い、上方又は側方から観察して液の色を比較する。
このとき試料溶液の呈する色は、比較液の呈する色より濃くてはならない。
(17) 水分定量法(カールフィッシャー法)
水分定量法は、メタノール及びピリジンの存在で、水がヨウ素及び二酸化イオウと次式に示すように定量的に反応することを利用して水分を定量する方法である。
H2O+I2+SO2+3C5H5N=2(C5H5N+H)I-+C5H5N・SO3
C5H5N・SO3+CH3OH=(C5H5N+H)O-SO2・OCH3
装置
通例、自動ビュレット2本、滴定フラスコ(250mL)、かき混ぜ器及び定電圧電流滴定装置からなる。
カールフィッシャー試液は吸湿性が非常に強いため、装置は、外部からの吸湿を防ぐようにする。防湿には、シリカゲル又は塩化カルシウム(水分測定用)等を用いる。
試薬・試液
カールフィッシャー用メタノール メタノール1,000mLにマグネシウム末5g(4.5~5.4g)を加え、水分吸収管(水分測定用塩化カルシウム)を付けた還流冷却器を取り付けて加熱し、必要ならば、塩化第二水銀0.1g(0.05~0.14g)を加えて反応を促進させる。ガスの発生が止んだ後、湿気を遮ってメタノールを蒸留し、湿気を避けて保存する。本品1mL中の水分は、0.5mg以下とする。
カールフィッシャー用ピリジン ピリジンに水酸化カリウム又は酸化バリウムを加え、密栓し、数日間放置した後、そのまま湿気を遮って蒸留し、湿気を避けて保存する。本品1mL中の水分は、1mg以下とする。
カールフィシャー試液
調製 ヨウ素63g(62.5~63.4g)をカールフィッシャー用ピリジン100mLに溶かし、氷冷し、乾燥二酸化イオウを通じ、その増量が32.3gに達したとき、二酸化イオウを通じることを止め、カールフィッシャー用メタノールを加えて500mLとし、24時間以上放置した後用いる。この試液は、日時の経過と共に変化するので、用時標定する。遮光して湿気を避け、冷所に保存する。
標定 操作法に従い、カールフィッシャー用メタノール25mLを乾燥滴定フラスコにとる。これをあらかじめカールフィッシャー試液で終点まで滴定して、フラスコ内を無水の状態にしておく。次に、水約50mgを0.1mgの桁まで量り、30秒以内に滴定フラスコに入れ、激しくかき混ぜながら、カールフィッシャー試液で終点まで滴定する。カールフィッシャー試液の1mLに対応する水(H2O)のmg数fを次式により求める。
f=水(H2Oの採取量(mg)/水(H2O)の滴定に要したカールフィッシャー試液の量(mL)
水・メタノール標準液
調製 カールフィッシャー用メタノール500mLを1,000mLの乾燥全量フラスコに入れ、水2.0mLを加え、カールフィッシャー用メタノールを標線まで加えて1,000mLとする。
この標準液の標定は、カールフィッシャー試液の標定に続いて行う。遮光して湿気を避け、冷所に保存する。
標定 操作法に従い、カールフィッシャー用メタノール25mLを乾燥滴定フラスコにとる。これをあらかじめカールフィッシャー試液で終点まで滴定して、フラスコ内を無水の状態にしておく。次に、全量ピペットを用いてカールフィッシャー試液10mLを加え、調製した水・メタノール標準液で終点まで滴定する。水・メタノール標準液1mL中の水(H2O)のmg数f′を次式により求める。
′=(f×10)/滴定に要した水・メタノールの標準液の量(mL)
操作法
カールフィッシャー試液による滴定は、湿気を避けて行い、原則として、これを標定したときの温度と同一の温度で行う。被滴定液中に2本の白金電極を浸し、可変抵抗器を適当に調節して一定の電流(5~10マイクロアンペア)を流しておき、カールフィッシャー試液を滴加した場合、滴定が進むにつれて、回路中のマイクロアンメーターの針が大きく振れ、数秒で再び元の位置に戻る。滴定の終点に達すると、マイクロアンメーターの振れ(50~150マイクロアンペア)が30秒間又はそれ以上の間持続する。この状態になったときを滴定の終点とする。ただし、逆滴定の場合、カールフィッシャー試液が過量に存在する間はマイクロアンメーターの針が振り切れ、終点に達すると急に元の位置に戻る。マイクロアンメーターの代わりにマジックアイ付電位差計を用いてもよい。
カールフィッシャー試液による滴定は、別に規定する場合を除き、次のいずれかの方法によるものとする。終点は、通例、逆滴定を行う場合の方が明瞭に判別できる。
① 直接滴定
別に規定する場合を除き、次の方法によるものとする。
カールフィッシャー用メタノール25mLを乾燥滴定フラスコに入れ、これをあらかじめカールフィッシャー試液で終点まで滴定して、フラスコ内を無水の状態にしておく。次に、水分10~50mgを含む量の試料を有効数字3桁まで量り、その数値を記録し、30秒以内に滴定フラスコに入れ、かき混ぜて溶かし、激しくかき混ぜながらカールフィッシャー試液で終点まで滴定する。試料が溶剤に溶けないときは、手早く粉末とし、その質量を量り、30秒以内に滴定フラスコに入れ、湿気を避けて30分間かき混ぜた後、激しくかき混ぜながら滴定を行う。
水(H2O)%=((試液の滴定に要したカールフィッシャー試液の量(mL)×f)/試料の量(mg))×100
② 逆滴定
別に規定する場合を除き、次の方法によるものとする。
カールフィッシャー用メタノール25mLを乾燥滴定フラスコに入れ、これをあらかじめカールフィッシャー試液で終点まで滴定し、フラスコ内を無水の状態にしておく。次に、水分10~50mgを含む量の試料を有効数字3桁まで量り、その数値を記録し、30秒以内に滴定フラスコに入れ、過量のカールフィッシャー試液の一定量を加え、かき混ぜて溶かし、激しくかき混ぜながら水・メタノール標準液で終点まで滴定する。試料が溶剤に溶けないときは、手早く粉末とし、その質量を量り、30秒以内に滴定フラスコに入れ、過量のカールフィッシャー試液の一定量を加え、湿気を避けて30分間かき混ぜた後、激しくかき混ぜながら滴定を行う。
水(H2O)%=((カールフィシャー試液の量(mL)×f-滴定に要した水・メタノール標準液の(mL)×f′)/試料の量(mg))×100
(18) 生菌剤試験法
生菌剤試験法は、微生物学的方法又は化学的方法により試料中の生菌の同定を行う試験法である。この試験に使用する水、試薬・試液及び計量器・用器は、必要に応じ無菌のものを用いる。
培地の種類並びにその組成及びpH
別に規定する場合を除き、次の表に掲げる組成及びpHを有するものを使用する。ただし、培地の成分として単に「ペプトン」と記載してある場合は、カゼイン製ペプトンを用いても差し支えない。培地のpHの調整は、1mol/L水酸化ナトリウム試液又は1mol/L塩酸試液を用い、滅菌後のpHが所定のものとなるようにする。
| 培地の組成及びpH | |||||||||||
| 培地1,000mLの組成 | 培地番号 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| カゼイン製ペプトン(g) | 5 | 20 | 20 | 10 | 5 | 17 | 10 | 15 | |||
| プロテオーゼペプトン(g) | 10 | 10 | |||||||||
| 大豆ペプトン(g) | 3 | 3 | 3 | 5 | |||||||
| ペプトン(g) | 10 | ||||||||||
| 肉エキス(g) | 2.4 | 10 | 10 | 5 | 5 | 2.2 | |||||
| 塩化ナトリウム(g) | 0.01 | 1.5 | 1.5 | 5 | 0.01 | 5 | 3 | 5 | 5 | ||
| 酵母エキス(g) | 5 | 2.2 | 2.2 | 5 | 5 | ||||||
| 肝臓エキス(g) | 3.2 | 1.2 | |||||||||
| ブドウ糖(g) | 10 | 2 | 2.5 | 10 | |||||||
| 乳糖(g) | 20 | 20 | |||||||||
| ポリソルベート80(mL) | 1 | 1 | 1 | ||||||||
| 可溶性デンプン(g) | 0.5 | ||||||||||
| リン酸二水素カリウム(g) | 1 | 0.5 | 2.5 | ||||||||
| リン酸一水素カリウム(g) | 1 | 0.5 | 2.5 | ||||||||
| 硫酸マグネシウム(g) | 0.2 | 0.3 | |||||||||
| 硫酸第一鉄(g) | 0.01 | 0.01 | |||||||||
| 硫酸マンガン(g) | 0.007 | 0.01 | |||||||||
| 硫酸亜鉛(g) | 0.001 | ||||||||||
| 硫酸コバルト(g) | 0.001 | ||||||||||
| 硫酸銅(g) | 0.001 | ||||||||||
| L-システイン塩酸塩一水和物(g) | 0.5 | 0.3 | |||||||||
| シリコン(g) | 0.2 | ||||||||||
| 消化血清(g) | 13.5 | ||||||||||
| チオグリコール酸ナトリウム(g) | 0.3 | ||||||||||
| 牛心臓抽出液(g) | 500 | ||||||||||
| トリプトース(g) | 10 | ||||||||||
| ブロムクレゾールパープル試液(mL) | 1 | ||||||||||
| カンテン(g) | 15 | 15 | 15 | 15 | 20 | 15 | |||||
| 水 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | 適量 | |
| 滅菌後のpH | 7.1~7.3 | 6.9~7.1 | 6.9~7.1 | 6.9~7.1 | 5.9~6.1 | 7.2~7.4 | 6.4~7.0 | 7.2~7.4 | 7.2~7.4 | 7.3~7.5 | |
注
1)1号培地及び9号培地は滅菌後、100mLに対して馬脱繊維血液5mLを加えたものを使用する。
2)5号培地のpH調整は、1mol/L水酸化ナトリウム試液又は希硫酸を用いる。
① 染色法
染色法は、生菌を適当な染色液を用いて染色し、生菌の物理的・化学的性質、形状及び芽胞の有無を判定する試験法である。
(ⅰ) グラム染色法 次の2つの方法のいずれか適当な方法を用いる。
ア HUCKERの変法
スライドグラス上に塗抹し、固定した試料にフッカーの染色液2滴を加え、30~60秒間放置して染色した後、スライドグラスを軽く振って液をきる。次に、ルゴール液を十分に加え、60秒間放置した後、水洗し、ろ紙で水を吸収する。スライドグラスを軽く動かしながら、脱色液として無水エタノール又はエタノール・アセトン混液(7:3)を用いて洗液がほぼ無色になるまで脱色する。その後、水洗し、ろ紙で水を吸収する。これにサフラニン溶液(1→200)2滴を加え、60秒間放置し、後染色(対比染色)した後、水洗し、乾燥する。
イ Lillieの変法
スライドグラス上に塗抹し、固定した試料にリリーの染色液2滴を加え、30秒間放置して染色した後、スライドグラスを軽く振って液をきる。次に、ヨウ素・ルゴール試液で数回洗った後、ヨウ素・ルゴール試液3滴を加え、30秒間放置する。ヨウ素・アセトン試液で十分に洗い流した後、ヨウ素・アセトン試液3滴を加え、30秒間放置する。その後、水洗し、ろ紙で水を吸収する。これに弱石炭酸フクシン液2滴を加え、30秒間放置し、後染色(対比染色)した後、水洗し、乾燥する。
(ⅱ) 芽胞染色法 WIRTZの法(SCHAEFFER-FULTONの変法)
スライドグラス上に塗抹し、固定した試料にマラカイトグリーン溶液(1→20)で1~3分間、加温又は加熱して染色した後、約30秒間水洗し、乾燥する。乾燥した塗抹面にサフラニン溶液(1→200)1~3滴を加え15~30秒間染色した後、水洗し、乾燥する。
② 糖分解能力試験法
糖分解能力試験法は、生菌を培養して培養液のpHを測定等することにより生菌の糖の分解能力の有無を判定する試験法である。
次の2つの方法のいずれか適当な方法を用いる。
(ⅰ) 第1法
各条に規定する培地に生じた集落を白金耳でとり、8号培地10mL及び各条に規定する糖の水溶液(3→10)をろ過滅菌した後、ろ液0.3mLを加えた8号培地10mLにそれぞれ接種し、36~38℃で7日間嫌気的に培養し、標準液及び試料溶液とする。
pH測定法により標準液及び試料溶液のpHを測定し、標準液のpHから試料溶液のpHを引いた値が1未満のときを陰性、1以上のときを陽性とする。
(ⅱ) 第2法
各条に規定する培地に生じた集落を白金耳でとり、各条に規定する糖の水溶液(3→10)をろ過滅菌した後、ろ液0.1mLを加えた10号培地3mLに接種し、36~38℃で7日間培養し、試料溶液とする。
別に、陰性対照として10号培地3mLに、陽性対照としてブドウ糖溶液(3→10)をろ過滅菌した後、ろ液0.1mLを加えた10号培地3mLに、それぞれ菌を接種し、同様の操作を行う。
試料溶液が青紫色を呈した場合を陰性と、黄色を呈した場合を陽性とする。同時に、陰性対照が青紫色を、陽性対照が黄色を呈することを確認する。
③ 乳酸生成能力試験法
乳酸生成能力試験法は、生菌を培養して培養液中の乳酸を測定することにより、生菌の乳酸の生成能力の有無を判定する試験法である。
試料溶液及び標準溶液の調製
別に規定する場合を除き、次の方法による。
各条に規定する試料原液又は培地に生じた集落を白金耳でとり、2号培地10mLに接種し、36~38℃で7日間培養し、試料原液とする。試料原液1.0mLを10mLの共栓付き試験管に入れ、硫酸(1→2)0.25mL及びエーテル2mLを加え、緩やかに20回転倒混和した後、静置し、上層をとり、別の10mLの共栓付き試験管に入れ、同量の水を加え、同様に操作し、下層をとり、これをメンブランフィルター(孔経0.45μm以下)でろ過し、試料溶液とする。
乳酸1.0mLに水を加えて100mLとし、これを標準原液とする。標準原液1.0mLに水を加えて100mLとし、これをメンブランフィルター(孔経0.45μm以下)でろ過し、標準液とする。
使用する水は、液体クロマトグラフ用に精製したものを用いる。
ただし、エンテロコッカス フェカーリス、エンテロコッカス フェシウム及びバチルス コアグランスは好気的に、ラクトバチルス アシドフィルス及びラクトバチルス サリバリウスは嫌気的に培養する。
操作法
試料溶液及び標準液各20μLにつき、次の条件で液体クロマトグラフ法により試験を行うとき、試料溶液から得たピークの保持時間は、標準液から得たピークの保持時間に一致する。
操作条件
検出器:紫外吸光光度計(測定波長:210nm)
カラム:内径4.6mm、長さ150mmのステンレス管に、粒径5μmの液体クロマトグラフ用オクタデシルシリル化シリカゲルを充填したもの
カラム温度:40℃
移動相:りん酸(1→1,000)
流量:毎分約1.0mL
④ 酪酸生成能力試験法
酪酸生成能力試験法は、生菌を培養して培養液中の酪酸を測定することにより、生菌の酪酸の生成能力の有無を判定する試験法である。
試料溶液及び標準溶液の調整
各条に規定する試料原液又は培地に生じた集落を白金耳でとり、7号培地10mLに接種し、36~38℃で7日間嫌気的に培養し、試料原液とする。試料原液1.0mLを10mLの共栓付き試験管に入れ、硫酸(1→2)0.25mL及びエーテル2mLを加え、緩やかに20回転倒混和した後、静置し、上層をとり、別の10mLの共栓付き試験管に入れ、同量の水を加え、同様に操作し、下層をとり、これをメンブランフィルター(孔経0.45μm以下)でろ過し、試料溶液とする。
酪酸1.0mLに水を加えて100mLとし、これを標準原液とする。標準原液1.0mLに水を加えて100mLとし、これをメンブランフィルター(孔経0.45μm以下)でろ過し、標準液とする。
使用する水は、液体クロマトグラフ用に精製したものを用いる。
操作法
試料溶液及び標準液各20μLにつき、次の条件で液体クロマトグラフ法により試験を行うとき、試料溶液から得たピークの保持時間は、標準液から得たピークの保持時間に一致する。
操作条件
検出器:紫外吸光光度計(測定波長:210nm)
カラム:内径4.6mm、長さ150mmのステンレス管に、粒径5μmの液体クロマトグラフ用オクタデシルシリル化シリカゲルを充填したもの
カラム温度:40℃
移動相:りん酸(1→1,000)
流量:毎分約1.0mL
(19) 生菌剤定量法
生菌剤定量法は、微生物学的方法により試料中の生菌の菌数の測定を行う試験法である。この試験に使用する水、試薬・試液及び計量器・用器は、必要に応じ無菌のものを用いる。
希釈液
希釈液は、次に掲げる組成及びpHを有するものを滅菌して使用する。
1号希釈液(pH7.0)
カゼカゼイン製ペプトン1g(0.5~1.4g)及び塩化ナトリウム5g(4.5~5.4g)に水約750mLを加えて溶かし、pHを6.9~7.1に調整した後、更に水を加えて1,000mLとする。
2号希釈液(pH7.0)
リン酸二水素カリウム4.5g(4.45~4.54g)、リン酸一水素ナトリウム12水塩6g(5.5~6.4g)、ポリソルベート80 0.5g(0.45~0.54g)、L-システイン塩酸塩一水和物0.5g(0.45~0.54g)及びカンテン0.5g(0.45~0.54g)に水約750mLを加えて溶かし、pHを6.9~7.1に調整した後、更に水を加えて1,000mLとする。
培地の種類並びにその組成及びpH
(18)の生菌剤試験法の項を準用する。
試料溶液の調製
別に規定する場合を除き、次の方法によるものとする。
本品約1gを0.001gの桁まで量り、その数値を記録し、全量ピペットを用いて希釈液50mLを加え、よく振り混ぜ、試料原液とする。この原液1mLを全量ピペットを用いて量り、別に、全量ピペットを用いて量った希釈液9mLに加え、10倍に希釈する。この操作を繰り返し、1mL中に生菌を30~300個を含む濃度又は300~3,000個含む濃度に調製し、試料溶液とする。なお、必要に応じて希釈時に界面活性剤を用いる。
操作法
試料溶液が1mL中に生菌を30~300個含む濃度の場合は、第1法を、1mL中に生菌を300~3,000個含む濃度の場合は、第2法を用いる。
① 第1法
試料溶液1mLずつを5枚のペトリ皿に入れ、これに50℃に保った試験用寒天培地を20mLずつ加え、30秒以内に混和し、固化させる。必要ならば、ペトリ皿の底に試験用寒天培地で基層を作り、上記操作を行った後、更に試験用寒天培地を加えて重層とする。これを36~38℃で各条に規定する期間培養して出現した集落を数え、平均集落数を求める。
試料1g中の生菌数=(平均集落数×希釈倍率×50)/試料採取量(g)
希釈倍率:10倍希釈法による希釈倍数
② 第2法
あらかじめ試験用寒天培地を20mLずつ加え、固化させた5枚のペトリ皿に、試料溶液0.1mLずつを入れて塗布する。これを各条に規定する温度で各条に規定する期間培養し、出現した集落数を数え、平均集落数を求める。
試料1g中の生菌数=(平均集落数×希釈倍率×500)/試料採取量(g)
希釈倍率:10倍希釈法による希釈倍数
(20) 赤外吸収スペクトル測定法
赤外吸収スペクトル測定法は、赤外線が試料を通過するときに吸収される度合いを、各波数について測定する方法である。赤外吸収スペクトルは、横軸に波数(cm-1)を、縦軸に通常透過率(%)又は吸光度をとったグラフで示される。赤外吸収スペクトルは、その物質の化学構造により定まる。したがって、種々の波数における吸収を測定して物質を確認し、又は定量することができる。
装置
複光束式赤外分光光度計を用いる。
あらかじめ分光光度計を調整した後、測定を行う。特に、透過率の直線性は20~80%の間で偏差が1%以内、透過率の再現性は2回繰り返し測定し±0.5%、波数の再現性は波数3,000cm-1付近で±5cm-1、1,000cm-1付近で±1cm-1以内とする。波数目盛りは、通例、ポリスチレン膜の3,060cm-1、1,601cm-1、1,029cm-1、907cm-1等の吸収帯を用いて補正する。
操作法
試料は、主な吸収帯の透過率が20~80%の範囲になるように、次のいずれかの方法により調製する。窓板は、塩化ナトリウム、臭化カリウム、臭化ヨウ化タリウム等を使用する。
① 臭化カリウム錠剤法
固体試料1~2mgをめのう製乳鉢で粉末とし、これに赤外用臭化カリウム100~200mgを加え、湿気を吸わないように注意し、速やかによくすり混ぜた後、錠剤成形器に入れ、0.67kPa以下の減圧下に錠剤の単位面積(cm2)当たり5,000~10,000kgの圧力を5~8分間加えて製錠し、測定する。
② 溶液法
各条に規定する方法で調製した試料溶液を、液体用固定セルに注入し、測定する。補償光路側には、使用した溶媒を置く。固定セルの厚さは、通例、0.1mm又は0.5mmとする。
③ ペースト法
固定試料をめのう製乳鉢で粉末とし、流動パラフィン等を加え、よく練り合わせ、空気が入らないように注意しながら2枚の窓板の間に挟んで測定する。
④ 液膜法
液体試料1~2滴を2枚の窓板の間に挟んで測定する。液層を厚くする必要がある場合は、アルミニウム箔等を2枚の窓板の間に挟み、その中に液体試料がたまるようにする。
⑤ 薄膜法
試料を、薄膜のまま又は各条に規定する方法により薄膜を調製した後、測定する。
⑥ 気体試料測定法
試料を、排気した5~10cmの長さの光路をもつ気体セルに各条に規定する圧力で入れ、測定する。必要に応じて1m以上の光路をもつ長光路セルを用いることができる。
⑦ ATR法
ATR(減衰全反射)プリズム面に試料を密着させ、その反射スペクトルを測定する。
(21) 旋光度測定法
旋光度測定法は、平面偏光が光学的活性物質又はその溶液を通過するとき、その偏光面が回転する角度を旋光計で測定する方法である。
旋光度αxtとは、特定の単色光x(波長又は名称で記載する。)を用い、温度t℃で測定したときの旋光度をいい、右旋性を+、左旋性を-の記号で示す。
比旋光度 〔α〕xtは、次の式で表わす。
〔α〕xt=100α/lc
t:測定時の温度
x:用いたスペクトルの特定の単色光の波長又は名称(D線を用いたときは、Dと記載する。)
α:偏光面を回転した角度
l:試料溶液の層、すなわち、測定に用いた測定管の長さ(mm)
c:溶液1mL中に存在する試料のg数である。液状試料を溶液としないでそのまま用いたときは、その密度である。ただし、別に規定する場合を除き、この密度の代わりに、その比重を用いる。
操作法
この測定は、別に規定する場合を除き、温度は20℃、層長は100mm、光線はナトリウムスペクトルのD線で行う。
(22) 粗脂肪定量法
粗脂肪定量法は、試料中の脂肪その他のエーテル可溶性物質を定量する方法である。
各条に、例えば、「20.0%以下(2g)」と規定するものは、本品約2gを0.001gの桁まで量り、その数値を記録し、次の操作法により粗脂肪を定量するとき、その量が本品1gにつき、200mg以下であることを示す。
操作法
別に規定する場合を除き、次の方法によるものとする。
試料2~5gを0.01gの桁まで量り、その数値を記録し、径約2.2cm、高さ約9cmの円筒ろ紙に入れ、その上に脱脂綿を少量ずつ数回に分け軽く押さえるようにして詰める。これを95~100℃の乾燥器中で2時間乾燥し、デシケーター(シリカゲル)で放冷した後、脂肪秤量瓶(あらかじめ95~100℃で乾燥し、デシケーターで放冷した後、質量を測定しておいたもの)を連結したソックスレー脂肪抽出装置に入れ、エーテルで抽出する。16時間抽出した後、円筒ろ紙を除き、脂肪秤量瓶中のエーテルを回収する。
脂肪秤量瓶を外して残りのエーテルを揮発させ、95~100℃で3時間乾燥し、デシケーターで放冷した後、その質量を0.01gの桁まで量り、その数値を記録する。
(23) 粗繊維定量法
粗繊維定量法は、試料を1.25%硫酸、1.25%水酸化ナトリウム液で順次処理し、セルロースその他の不溶又は難溶性の成分を定量する方法である。
各条に、例えば、「5.0%以下(2g)」と規定するものは、本品約2gを0.001gの桁まで量り、その数値を記録し、次の操作法により粗繊維を定量するとき、その量が本品1gにつき、50mg以下であることを示す。
操作法
別に規定する場合を除き、次の方法によるものとする。
試料2~5gを0.001gの桁まで量り、その数値を記録し、500mLのトールビーカーに入れ、5%硫酸溶液50mLを加え、さらに、水を加えて200mLとし、静置した液面に沿ってトールビーカーの外壁に標線を付しておく。次に、トールビーカーを時計皿又は冷却器で覆い、30分間激しく煮沸する。その間蒸発する水分は、常時熱水を補って硫酸の濃度を1.25%に保つ。30分経過後、酸不溶解物を0.044mmのステンレス金網(325号のふるい又はこれに相当するもの)でろ過し、酸性を呈しなくなるまで熱水で洗浄する。
酸不溶解物を水130~140mLを用いて元のトールビーカーに移し、5%水酸化ナトリウム液50mLを加え、標線まで水を加えて200mLとし、時計皿又は冷却器で覆い30分間激しく煮沸した後、ろ紙(あらかじめ秤量皿に入れ、135±2℃で2時間乾燥し、デシケーター(シリカゲル)で放冷した後、質量を測定しておいたもの)でろ過し、ろ液がアルカリ性を呈しなくなるまで熱水で洗浄し、アルコール、エーテルの順にそれぞれ3~4回洗浄した後、3~4時間風乾する。
次に、この酸・アルカリ不溶解物をろ紙と共に前の秤量皿に移し、135±2℃で2時間乾燥し、デシケーターで放冷した後、その質量を0.001gの桁まで量り、記録しておく。
その後、これをるつぼ(あらかじめ強熱し、デシケーターで放冷した後、質量を測定しておいたもの)に入れ、穏やかに加熱して炭化させた後、550~600℃で2時間灰化し、デシケーターで放冷した後、その質量を0.001gの桁まで量って粗灰分の量を求める。酸・アルカリ不溶解物の量から粗灰分の量を控除し、粗繊維の量とする。
(24) 窒素定量法
窒素定量法は、窒素を含む有機化合物を硫酸で分解し、硫酸アンモニウムとし、そのアンモニアを定量する方法である。
各条に、例えば、「5.0~7.0%(ケルダール法)」と規定するものは、窒素約20~30mgを含む量の本品を有効数字3桁まで量り、その数値を記録し、次のケルダール法により窒素を定量するとき、その量が本品1gにつき、50~70mgであることを示す。
ケルダール法
装置
図に示すものを用いる。総硬質ガラス製で、接続部をすり合わせにすることができる。装置に用いるゴムは、全て1mol/L水酸化ナトリウム試液中で10~30分間煮沸し、次に、水中で30~60分間煮沸し、最後に、水でよく洗ってから用いる。
| A:ケルダールフラスコ |  |
| B:ガラス管 | |
| C:アルカリ溶液注入用漏斗 | |
| D,E:ピンチコック付ゴム管 | |
| F:しぶき止め | |
| G:蒸留管 | |
| H:冷却管 | |
| J:受器 |
操作法
別に規定する場合を除き、次の方法によるものとする。
窒素(N)20~30mgを含む量の試料を有効数字3桁まで量り、その数値を記録し、ケルダールフラスコAに入れ、これに硫酸カリウムの粉末5g(4.5~5.4g)、硫酸銅0.5g(0.45~0.54g)及び硫酸20mLを加える。次に、フラスコを約45°に傾け、泡立ちがほとんど止むまで静かに加熱し、更に温度を上げて沸騰させ、内容物が青色で、透明な溶液となった後、更に1~2時間加熱する。
放冷した後、適量の水を徐々に加え、放冷し、あらかじめ水蒸気を通じて洗った蒸留装置にフラスコを連結する。
受器Jには、ホウ酸溶液(1→25)20mL及びブロムクレゾールグリーン・メチルレッド試液3滴を入れ、適量の水を加え、冷却器Hの下端をこの溶液に浸す。次に、漏斗Cから水酸化ナトリウム溶液(2→5)85mLを徐々に加え、さらに、少量の水で洗い込み、Dの部分のピンチコックを閉じ、ケルダールフラスコを軽く揺り動かして内容物を混和した後、水蒸気を通じて留液約120mLを得るまで蒸留する。その後、冷却器の下端を受器の液面から離し、更にしばらく蒸留を続けた後、冷却器の下端を少量の水で洗い込み、0.05mol/L硫酸で滴定する。この場合において、滴定の終点は、溶液の緑色が微灰色を経て微灰赤紫色に変わるときとする。
同様の方法で空試験を行い補正する。
0.05mol/L硫酸1mL=1.401mgN
セミミクロケルダール法
装置
図に示すものを用いる。総硬質ガラス製で、接続部をすり合わせにすることができる。装置に用いるゴムは、全て1mol/L水酸化ナトリウム試液中で10~30分間煮沸し、次に、水中で30~60分間煮沸し、最後に、水でよく洗ってから用いる。
| A:ケルダールフラスコ |  |
| B:水蒸気発生器で、硫酸2~3滴を加えた水を入れ、突沸を避けるために沸騰石を入れる。 | |
| C:しぶき止め | |
| D:給水用漏斗 | |
| E:蒸気管 | |
| F:アルカリ溶液注入用漏斗 | |
| G:ピンチコック付ゴム管 | |
| H:小孔(径は管の内径にほぼ等しい。) | |
| J:冷却器(下端は斜めに切ってある。) | |
| K:受器 |
操作法
別に規定する場合を除き、次の方法によるものとする。
窒素(N:14.01)2~3mgを含む量の試料を有効数字3桁まで量り、その数値を記録し、又はピペットで量り、ケルダールフラスコAに入れる。別に、硫酸カリウム10g(9.5~10.4g)及び硫酸銅1g(0.5~1.4g)を混合して粉末とし、その1g(0.5~1.4g)をフラスコに加え、フラスコの首に付着した試料を少量の水で洗い込み、さらに、フラスコの内壁に沿って硫酸7mLを加える。
次に、フラスコを振り動かしながら、強過酸化水素水1mLを少量ずつ内壁に沿って注意して加える。フラスコを加熱し、溶液が青色で、澄明となり、フラスコの内壁に炭化物を認めなくなったとき、加熱を止める。必要ならば、冷却した後、強過酸化水素水少量を追加し、再び加熱する。放冷した後、水20mLを注意しながら加え、冷却する。フラスコを、あらかじめ水蒸気を通じて洗った蒸留装置に連結する。受器Kには、ホウ酸溶液(1→25)15mL及びブロムクレゾールグリン・メチルレッド試液3滴を入れ、適量の水を加え、冷却器Jの下端をこの溶液に浸す。漏斗Fから水酸化ナトリウム溶液(2→5)30mLを加え、注意して水10mLで洗い込み、30秒以内にピンチコック付ゴム管Gのピンチコックを閉じ、水蒸気を通じて留液80~100mLを得るまで蒸留する。冷却器Jの下端を液面から離し、少量の水でその部分を洗い込み、0.005mol/L硫酸で滴定する。この場合において、滴定の終点は、溶液の緑色が微灰青色を経て微灰赤紫色に変わるときとする。
0.005mol/L硫酸1mL=0.1401mgN
(25) 定性反応
定性反応は、試料の確認試験に用い、通例、その溶液2~5mLを量り、試験を行う。
亜鉛塩
① 亜鉛塩の中性~アルカリ性溶液に硫化アンモニウム試液又は硫化ナトリウム試液を加えるとき、帯白色の沈殿を生じる。沈殿を分取し、これに希酢酸を加えても溶けないが、希塩酸を追加するとき、溶ける。
② 亜鉛塩の溶液にフェロシアン化カリウム試液を加えるとき、白色の沈殿を生じ、この一部に希塩酸を追加しても、沈殿は溶けない。また、残りの沈殿の一部に1mol/L水酸化ナトリウム試液を追加するとき、溶ける。
③ 亜鉛塩の溶液にリン酸を加えて酸性とし、硫酸銅溶液(1→1,000)1滴及びチオシアン酸水銀アンモニウム試液2mLを加えるとき、淡紫色の沈殿を生じる。
アルミニウム塩
① アルミニウム塩の溶液に塩化アンモニウム試液及びアンモニア試液を加えるとき、白色のゲル状の沈殿を生じ、過量のアンモニア試液を追加しても、沈殿は溶けない。
② アルミニウム塩の溶液に1mol/L水酸化ナトリウム試液を加えるとき、白色のゲル状の沈殿を生じ、過量の1mol/L水酸化ナトリウム試液を追加するとき、沈殿は溶ける。
③ アルミニウム塩の溶液に硫化ナトリウム試液を加えるとき、白色のゲル状の沈殿を生じ、過量の硫化ナトリウム試液を追加するとき、沈殿は溶ける。
④ アルミニウム塩の溶液に白色のゲル状の沈殿が生じるまでアンモニア試液を加え、アリザリンレッドS試液5滴を追加するとき、沈殿は、赤色に変わる。
アンモニウム塩
アンモニウム塩に過量の1mol/L水酸化ナトリウム試液を加え、加温するとき、アンモニア臭のガスを発し、このガスは、潤した赤色リトマス紙を青変する。
塩化物
① 塩化物の溶液に硫酸及び過マンガン酸カリウムを加えて加熱するとき、塩素臭のガスを発し、このガスは、潤したヨウ化カリウムデンプン紙を青変する。
② 塩化物の溶液に硝酸銀試液を加えるとき、白色の沈殿を生じる。沈殿を分取し、この一部に希硝酸を加えても溶けない。また、残りの沈殿の一部に過量のアンモニア試液を加えるとき、溶ける。
カリウム塩
① カリウム塩につき、炎色反応を行うとき、淡紫色を呈する。炎が黄色のときは、コバルトガラスを通して観察すると赤紫色に見える。
② カリウム塩の中性溶液に酒石酸水素ナトリウム試液を加えるとき、白色の結晶性の沈殿を生じる。沈殿の生成を速くするには、ガラス棒で試験管の内壁をこする。沈殿を分取し、これにアンモニア試液、1mol/L水酸化ナトリウム試液又は炭酸ナトリウム試液を加えるとき、いずれも溶ける。
③ カリウム塩の酢酸酸性溶液にコバルト亜硝酸ナトリウム試液を加えるとき、黄色の沈殿を生じる。
④ カリウム塩に過量の1mol/L水酸化ナトリウム試液を加え、加温しても、アンモニアの臭いを発しない(アンモニウム塩との区別)。
カルシウム塩
① カルシウム塩につき、炎色反応を行うとき、黄赤色を呈する。
② カルシウム塩の溶液に炭酸アンモニウム試液を加えるとき、白色の沈殿を生じる。
③ カルシウム塩の溶液にシュウ酸アンモニウム試液を加えるとき、白色の沈殿を生じる。沈殿を分取し、これに希酢酸を加えても溶けないが、希塩酸を追加するとき、溶ける。
④ カルシウム塩の中性溶液にクロム酸カリウム試液10滴を加えて加熱しても、沈殿を生じない(ストロンチウム塩との区別)。
クエン酸塩
① クエン酸塩の溶液に過量の硫酸第二水銀試液を加えて沸騰するまで加熱し、過マンガン酸カリウム試液を加えるとき、脱色し、白色の沈殿を生じる。沈殿を分取し、これに塩化ナトリウム試液を加えるとき、溶ける。
② クエン酸塩の中性溶液に等容量の希硫酸を加え、その2/3容量の過マンガン酸カリウム試液を加え、試液の色が消えるまで加熱した後、全量の1/10容量の臭素試液を滴加するとき、白色の沈殿を生じる。
③ クエン酸塩の中性溶液に過量の塩化カルシウム試液を加え、煮沸するとき、白色の結晶性の沈殿を生じる。沈殿を分取し、この一部に1mol/L水酸化ナトリウム試液を加えても溶けない。また、他の一部に希塩酸を加えるとき、溶ける。
コハク酸塩
コハク酸塩の溶液のpHを6~7に調整し、この溶液に塩化第二鉄試液1mLを加えるとき、褐色の沈殿を生じる。
酢酸塩
① 酢酸塩に、硫酸(1→2)を加え、加温するとき、酢酸の臭いを発する。
② 酢酸塩に硫酸及び少量のエタノールを加えて加熱するとき、酢酸エチルの臭いを発する。
③ 酢酸塩の中性溶液に塩化第二鉄試液を加えるとき、溶液は赤褐色を呈し、煮沸するとき、赤褐色の沈殿を生じる。これに塩酸を追加するとき、沈殿は溶け、溶液の色は黄色に変わる。
酒石酸塩
① 酒石酸塩の中性溶液に硝酸銀試液を加えるとき、白色の沈殿を生じる。沈殿を分取し、この一部に硝酸を加えるとき、沈殿は溶ける。また、残りの沈殿の一部にアンモニア試液を加え、加温するとき、沈殿は溶け、徐々に器壁に銀鏡を生じる。
② 酒石酸塩の溶液に酢酸2滴、硫酸第一鉄試液1滴及び過酸化水素試液2~3滴を加え、さらに、過量の1mol/L水酸化ナトリウム試液を加えるとき、赤紫色~紫色を呈する。
③ 酒石酸塩の溶液2~3滴に、あらかじめレゾルシン溶液(1→50)2~3滴及び臭化カリウム溶液(1→10)2~3滴を加えた硫酸5mLを加え、水浴上で5~10分間加熱するとき、濃青色を呈する。これを冷却した後、過量の水中に注ぐとき、溶液は、赤色を呈する。
硝酸塩
① 硝酸塩の溶液に等容量の硫酸を混和し、冷却した後、硫酸第一鉄試液を層積するとき、接界面に暗褐色の輪帯を生じる。
② 硝酸塩の溶液にジフェニルアミン試液を加えるとき、溶液は、青色を呈する。
③ 硝酸塩の硫酸酸性溶液に過マンガン酸カリウム試液を加えても、試液の赤紫色は、退色しない(亜硝酸塩との区別)。
炭酸塩
① 炭酸塩に希塩酸を加えるとき、泡立ってガスを発生する。このガスを水酸化カルシウム試液中に通じるとき、30秒以内に白色の沈殿を生じる(炭酸水素塩と共通)。
② 炭酸塩の溶液に硫酸マグネシウム試液を加えるとき、白色の沈殿を生じ、希酢酸を追加するとき、沈殿は溶ける。
③ 炭酸塩の冷溶液にフェノールフタレイン試液1滴を加えるとき、溶液は、赤色を呈する(炭酸水素塩との区別)。
炭酸水素塩
① 炭酸水素塩に希塩酸を加えるとき、泡立ってガスを発生する。このガスを水酸化カルシウム試液中に通じるとき、30秒以内に白色の沈殿を生じる(炭酸塩と共通)。
② 炭酸水素塩の溶液に硫酸マグネシウム試液を加えるとき、沈殿を生じないが、煮沸するとき、白色の沈殿を生じる。
③ 炭酸水素塩の冷溶液にフェノールフタレイン試液1滴を加えるとき、溶液は、赤色を呈しない、又は赤色を呈しても、極めて薄い(炭酸塩との区別)。
鉄塩、第一
① 第一鉄塩の弱酸性溶液にフェリシアン化カリウム試液を加えるとき、青色の沈殿を生じ、希塩酸を追加しても、沈殿は溶けない。
② 第一鉄塩の溶液に1mol/L水酸化ナトリウム試液を加えるとき、灰緑色のゲル状の沈殿を生じ、硫化ナトリウム試液を追加するとき、黒色の沈殿に変わる。沈殿を分取し、これに希塩酸を加えるとき、溶ける。
③ 第一鉄塩の中性又は弱酸性溶液にo -フェナントロリンのエタノール溶液(1→50)を滴加するとき、濃赤色を呈する。
鉄塩、第二
① 第二鉄塩の弱酸性溶液にフェロシアン化カリウム試液を加えるとき、青色の沈殿を生じ、希塩酸を追加しても、沈殿は溶けない。
② 第二鉄塩の溶液に1mol/L水酸化ナトリウム試液を加えるとき、赤褐色のゲル状の沈殿を生じ、硫化ナトリウム試液を追加するとき、黒色の沈殿に変わる。沈殿を分取し、これに希塩酸を加えるとき、溶け、溶液は白濁する。
③ 第二鉄塩の中性又は弱酸性溶液にチオシアン酸アンモニウム試液を加えるとき、溶液は、赤色を呈し、塩酸を追加してもこの色は消えないが、さらに、塩化第二水銀液を追加するとき、消える。
銅塩、第二
① 第二銅塩の塩酸酸性溶液によく磨いた板状の鉄を入れるとき、その表面に赤色の金属の膜を生じる。
② 第二銅塩の溶液に少量のアンモニア試液を加えるとき、淡青色の沈殿を生じ、過量のアンモニア試液を追加するとき、沈殿は溶け、溶液は濃青色を呈する。
③ 第二銅塩の溶液にフェロシアン化カリウム試液を加えるとき、赤褐色の沈殿を生じ、この一部に希硝酸を追加しても、沈殿は溶けない。また、他の一部にアンモニア試液を追加するとき、沈殿は溶け、溶液は濃青色を呈する。
④ 第二銅塩の溶液に硫化ナトリウム試液を加えるとき、黒色の沈殿を生じる。沈殿を分取し、この一部に希塩酸、希硫酸又は1mol/L水酸化ナトリウム試液を加えても溶けない。また、残りの沈殿の一部に熱希硝酸又はシアン化カリウム試液を加えるとき、溶ける。
ナトリウム塩
① ナトリウム塩につき、炎色反応を行うとき、黄色を呈する。
② ナトリウム塩の中性又は弱アルカリ性濃溶液にピロアンチモン酸カリウム試液を加えるとき、白色の結晶性の沈殿を生じる。沈殿の生成を速くするには、ガラス棒で試験管の内壁をこする。
乳酸塩
乳酸塩の硫酸酸性溶液に過マンガン酸カリウム試液を加えて加熱するとき、アセトアルデヒドの臭いを発する。
芳香族アミン、第一
芳香族第一アミンの酸性溶液に氷冷しながら亜硝酸ナトリウム試液3滴を加え、振り混ぜ、2分間放置し、次に、スルファミン酸アンモニウム試液1mLを加え、よく振り混ぜ、1分間放置した後、シュウ酸N-(1-ナフチル)-N′-ジエチルエチレンジアミン試液1mLを加えるとき、溶液は、赤紫色を呈する。
マグネシウム塩
① マグネシウム塩の溶液に炭酸アンモニウム試液を加えるとき、白色の沈殿を生じ、塩化アンモニウム試液を追加するとき、沈殿は溶ける。さらに、リン酸一水素ナトリウム試液を追加するとき、白色の結晶性の沈殿を生じる。
② マグネシウム塩の溶液に1mol/L水酸化ナトリウム試液を加えるとき、白色のゲル状の沈殿を生じ、更に過量の1mol/L水酸化ナトリウム試液を加えても沈殿は溶けないが、ヨウ素試液を追加するとき、沈殿は、暗褐色に染まる。
マンガン塩
① マンガン塩の溶液にアンモニア試液を加えるとき、白色の沈殿を生じる。この一部に硝酸銀試液を追加するとき、沈殿は、黒色に変わる。また、残りの沈殿の一部を放置するとき、沈殿の上部が褐色を帯びてくる。
② マンガン塩の希硝酸酸性溶液に少量のビスマス酸ナトリウムの粉末を加えるとき、溶液は、赤紫色を呈する。
ヨウ化物
① ヨウ化物の溶液に硝酸銀試液を加えるとき、黄色の沈殿を生じる。この一部に希硝酸を、また、残りの沈殿の一部に強アンモニア水を追加しても、いずれも沈殿は溶けない。
② ヨウ化物の酸性溶液に亜硝酸ナトリウム試液1~2滴を加えるとき、溶液は黄褐色を呈し、その後、黒紫色の沈殿を生じる。デンプン試液を追加するとき、溶液は、濃青色を呈する。
硫酸塩
① 硫酸塩の溶液に塩化バリウム試液を加えるとき、白色の沈殿を生じ、希硝酸を追加しても、沈殿は溶けない。
② 硫酸塩の中性溶液に酢酸鉛試液を加えるとき、白色の沈殿を生じ、酢酸アンモニウム試液を追加するとき、沈殿は溶ける。
③ 硫酸塩の溶液に等容量の希塩酸を加えても白濁せず(チオ硫酸塩との区別)、二酸化イオウの臭いを発しない(亜硫酸塩との区別)。
リン酸塩(正リン酸塩)
① リン酸塩の中性溶液に硝酸銀試液を加えるとき、黄色の沈殿を生じ、希硝酸又はアンモニア試液を追加するとき、沈殿は溶ける。
② リン酸塩の中性又は希硝酸酸性溶液にモリブデン酸アンモニウム試液を加え、加温するとき、黄色の沈殿を生じ、1mol/L水酸化ナトリウム試液又はアンモニア試液を追加するとき、沈殿は溶ける。
③ リン酸の中性又はアンモニアアルカリ性溶液にマグネシア試液を加えるとき、白色の結晶性の沈殿を生じ、希塩酸を追加するとき、沈殿は溶ける。
(26) 鉛試験法
鉛試験法は、試料中に混在する鉛の量を試験する方法である。
(ⅰ) ジチゾン法
試液・標準液
クエン酸アンモニウム溶液 クエン酸アンモニウム45g(44.5~45.4g)に水を加えて溶かし、100mLとする。
亜硫酸ナトリウム溶液 亜硫酸ナトリウム15g(14.5~15.4g)に水を加えて溶かし、100mLとする。用時調製する。
シアン化カリウム溶液 シアン化カリウム10g(9.5~10.4g)に水を加えて溶かし、100mLとする。
希シアン化カリウム溶液 シアン化カリウム溶液10mLに水を加えて100mLとする。用時調製する。
ジチゾン・ベンゼン溶液 ジチゾンを乳鉢中でよくすりつぶし、その0.05g(0.045~0.054g)にクロロホルム100mLを加えて溶かした後、分液漏斗に入れ、強アンモニア水(1→100)100mLずつで3回抽出する。全抽出液を合わせ、ベンゼン200mLずつで3回洗う。水層に希塩酸を加えて僅かに酸性とした後、ベンゼン200mLずつで2回抽出する。ベンゼン抽出液を合わせ、ベンゼンを加えて全量を約1,000mLとし、原液とする。原液をベンゼンで10倍に希釈した溶液につき、ベンゼンを対照液として、層長10mmで620nm付近における吸収の極大波長で吸光度Aを測定する。原液20,000/(70.6×A)mLを量り、1,000mLの全量フラスコに入れ、ベンゼンを標線まで加えて1,000mLとする。この溶液1,000mLは、ジチゾン(C13H12N4S)20mgを含む。用時調製する。
ジチゾン用鉛標準液 鉛標準液10mLを正確に量り、薄めた硝酸(1→100)を加えて正確に100mLとする。この液1mLは鉛(Pb)0.001mgを含む。用時製する。
操作法
各条に規定する量の試料溶液及び本品を用いないで試料溶液の場合と同様に操作して得た空試験液を量り、クエン酸アンモニウム溶液2mL及びメチルレッド試液2滴を加え、溶液が黄色を呈するまで強アンモニア水を滴加し、さらに、水を加えて全量を約100mLとする。これにシアン化カリウム溶液10mL及び亜硫酸ナトリウム溶液10mLを加え、よく振り混ぜ、水浴上で10~15分間加熱する。放冷した後、強アンモニア水1.5mLを加え、分液漏斗に移し、全量ピペットを用いてジチゾン・ベンゼン溶液10mLを加え、1分間強く振り混ぜ、水層を除く。さらに、希シアン化カリウム溶液40mLを加え、30秒間強く振り混ぜ、放置した後、ベンゼン層を分取し、ベンゼンを対照として、層長10mmで525nm付近における吸収の極大波長で吸光度AT及びABを測定する。同時にジチゾン用鉛標準液10mL及び水10mLについて、試料溶液と同様に操作し、吸光度AS及びA0を測定する。
鉛(Pb)の量(μg/g)=10×((AT-AB)/(AS-A0))×(試料溶液全量(mL)/試料溶液採取量(mL))×(1/試料の量(g))
注意:試験に用いる試薬及び試液は、鉛を含まず、又はほとんど含まないものを用いる。また、ガラス器具は、あらかじめ塩酸(1→2)でよく洗い、さらに、水で洗ったものを用いる。
(ⅱ) 原子吸光光度法
操作法
① 第1法
試料溶液及び標準液の調製
別に規定するものを除き、次の方法により試料溶液を調製する。
別に規定する量の試料を量り、白金製又は石英製のるつぼに入れ、硫酸少量を加えて潤し、徐々に加熱してできる限り低温でほとんど灰化した後、放冷し、更に硫酸1mLを加え、徐々に加熱して450~550℃で灰化するまで強熱する。残留物に少量の硝酸(1→150)を加えて溶かし、10mLの全量フラスコに入れ、更に硝酸(1→150)を標線まで加えて10mLとし、試料溶液とする。
また、別に規定するものを除き、鉛標準液1.0mLを全量ピペットを用いて量り、10mLの全量フラスコに入れ、硝酸(1→150)を標線まで加えて10mLとし、標準液とする。
試験
別に規定するものを除き、試料溶液及び標準液につき、原子吸光光度法(フレーム方式)により次の条件で吸光度を測定するとき、試料溶液の吸光度は、標準液の吸光度以下とする。
操作条件
光源ランプ 鉛中空陰極ランプ
分析線波長 283.3nm
支燃性ガス 空気
可燃性ガス アセチレン
② 第2法
試料溶液の調製
別に規定するものを除き、次の方法により試料溶液を調製する。
別に規定する量の試料を量り、ポリテトラフルオロエチレン製分解容器に入れ、硝酸0.5mLを加えて溶かした後、密封し、150℃で5時間加熱する。放冷した後、5mLの全量フラスコに入れ、水を標線まで加えて5mLとし、試料溶液とする。
試験
別に規定するものを除き、次の方法により試験を行う。
試料溶液3個以上をとり、原子吸光光度法(フレームレス方式(電気加熱方式))の標準添加法により次の条件で試験を行う。ただし、標準液は、鉛標準液適量を全量ピペットを用いて量り、水を加えて調製する。また、測定用溶液には、同容積の硝酸パラジウム試液を加え、よく混ぜ合わせる。硝酸10mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、水を標線まで加えて100mLとした溶液を用いて空試験を行い補正する。
操作条件
光源ランプ 鉛中空陰極ランプ
分析線波長 283.3nm
乾燥温度 110℃
灰化温度 600℃
原子化温度 2,100℃
(27) バイオオートグラフ法
バイオオートグラフ法は、ろ紙クロマトグラフ法又は薄層クロマトグラフ法を応用して分離した混合物中の力価を有する成分を、生物学的方法により、確認又はそのおおよその量を測定する方法とする。
操作法
ろ紙、薄層板、培養箱の形状、展開溶媒、常用標準希釈液、試料液、培地、試験菌液(又は試験胞子液)の調製、Rf値、移動距離、測定値、計算及び判定は、各条に規定する。
滅菌した培養箱に、培地及び試験菌液(又は試験胞子液)を加え、平板を作成する。これらの操作は、できる限り無菌的に行う。
ろ紙又は薄層板の原線を等間隔に4等分し、4区画になるよう垂直線を引く。各区画の原線の中央を原点とし、第1の区画から第3の区画までの各原点ごとに、各常用標準希釈液の高濃度のものから、順次5μLずつ、第4の区画の原点には、試料液5μLを、それぞれマイクロピペットを用いて点滴し、しみこませた後、風乾する。
このろ紙又は薄層板を、必要ならば、展開溶媒の気体で飽和した装置の中で30~60分間放置した後、展開溶媒を下降又は上昇させる。温度は、20~30℃とする。溶媒の下達線又は上達線が、ろ紙又は薄層板の下端又は上端より10~30mmに達したとき、展開を止め、ろ紙又は薄層板を取り出し、室温で放置し、溶媒を乾燥除去する。乾燥した後、ろ紙又は薄層板を区画線に沿って4等分し、必要ならば、不要部分を切り捨て、培養箱の培地上に、4切片のろ紙又は薄層板をそれぞれ約15mmの間隔で並べる。この際、ろ紙又は薄層板の各部と培地は、完全に接着するように注意して並べる。5~15分間接触させた後、ろ紙又は薄層板を取り去る。これらの操作は、雑菌が入らないよう注意して行う。培養箱は、32~37℃で17~20時間培養する。
(28) 薄層クロマトグラフ法
薄層クロマトグラフ法は、適当な固定相で作られた薄層を用い、混合物を移動相で展開させてそれぞれの成分に分離する方法であり、物質の確認又は純度の試験等に用いる。
薄層板の調製
通例、次の方法によるものとする。
薄層板は、50mm×200mm又は200mm×200mmの平滑で均一な厚さのガラス板に適当な器具を用いて、各条に規定する固定相固体の粉末を水で懸濁し、0.2~0.3mmの均一な厚さに塗布する。風乾した後、105~120℃の間の一定温度で30~60分間加熱、乾燥して調製する。ガラス板の代わりに適当なプラスチック板を使うことができる。薄層板は、湿気を避けて保存する。
操作法
別に規定する場合を除き、次の方法による。
薄層板の下端から約20mmの高さを原線とし、両側から少なくとも10mm離し、原線上に各条に規定する量の試料溶液及び標準液を、マイクロピペットを用いて約10mmの間隔でスポットし、風乾する。次に、この薄層板を展開用容器に入れて密閉し、常温で展開を行う。展開用容器には、あらかじめ展開溶媒を10mmの深さに入れ、展開溶媒の蒸気で飽和しておく。
展開溶媒の先端が原線から各条に規定する距離まで上昇したとき、薄層板を取り出し、風乾した後、各条に規定する方法により、それぞれのスポットの位置及び色等を調べる。Rf値は、次式により求める。
Rf=原線からスポットの中心までの距離/原線から溶媒先端までの距離
(29) pH測定法
pHを測定するためには、ガラス電極によるpH計を用いる。
pHは、次式で定義される値である。この値は、厳密な意味での物理化学的な意義をもつものではないが、試料の水溶液中の水素イオン濃度を、その逆数の常用対数で示した値とよく一致する。
pH=pHS+(E-ES)/α
pHS:pH標準液のpH値
E:試料溶液中でガラス電極と比較電極を組み合わせた電池の起電力
ES:pH標準液中でガラス電極と比較電極を組み合わせた電池の起電力
α=(2.3026×RT)/F
R:気体定数
T:絶対温度
F:ファラデー定数
装置
pH計は、通例、ガラス電極及び比較電極からなる検出部と、検出されたpHを指示する指示部からなる。指示部には、非対称電位調整用つまみ及び温度補償用つまみがある。温度補償用つまみのないものには、温度補償用感温部がある。
pH計は、次の操作法に従い、任意の一種類のpH標準液のpHを5回繰り返し測定するとき、その再現性が±0.05以内のものを用いる。なお、検出部は、毎回、測定の前に水でよく洗う。
操作法
ガラス電極を、あらかじめ水に数時間浸しておく。pH計は、電源を入れ、5分以上経ってから使用する。検出部をよく水で洗い、付着した水をろ紙等で軽く拭きとる。温度補償用つまみのあるものは、そのつまみをpH標準液の温度と一致させ、検出部を試料溶液のpH値に近いpH標準液中に浸し、2分以上経ってから、pH計の指示がその温度におけるpH標準液のpHになるように調整する。2点で調整する場合は、通例、リン酸塩pH標準液及び試料溶液のpH値に近いpH標準液を用いて、前記に準じて操作する。次に、検出部をよく水で洗い、付着した水をろ紙等で軽く拭きとった後、試料溶液に浸し、測定値を読み取る。
(30) 比重測定法
比重とは、物質の質量とそれと等体積の標準物質の質量の比をいう。
比重dtt’とは、温度t′℃における試料と温度t℃における水(H2O)との等体積の質量の比をいう。別に規定する場合を除き、測定には、第1法又は第2法を用い、数値に「約」を付記してあるときは、第3法を用いることができる。
操作法
① 第1法 比重瓶による測定法
比重瓶は、通例、内容10~100mLのガラス製容器で、温度計付きのすり合わせの栓と標線及びすり合わせの蓋のある側管とからなる。あらかじめ清浄かつ乾燥した比重瓶の質量Wを量る。次に、栓及び蓋を除き、試料を満たして規定温度t′℃より1~3℃低くし、泡が残らないように注意して栓をする。徐々に温度を上げ、温度計が規定温度を示したとき、標線の上部の試料を側管から除き、側管に蓋をし、外部をよく拭いた後、質量W1を量る。同じ比重瓶で水を用いて同様に操作し、その規定温度t℃におけるW2を量る。
dtt’=(W1-W)/(W2-W)
② 第2法 シュプレンゲル・オストワルドピクノメーターによる測定法
シュプレンゲル・オストワルドピクノメーターは、通例、内容1~10mLのガラス製容器で、図のように、両端は、肉厚細管(内径1~1.5mm、外径3~4mm)とし、一方の細管Aには、標線Cがあるものとする。あらかじめ清浄かつ乾燥したピクノメーターを、白金又はアルミニウム等の線Dで化学はかりの腕の鉤にかけ、質量Wを量る。次に、規定温度より3~5℃低い試料中に細管Bを浸す。Aには、ゴム管又はすり合わせの細管を付け、泡が入らないように注意し、試料をCの上まで吸い上げる。その後、規定温度t′℃の水浴中に約15分間浸した後、Bの端にろ紙片をあて、試料の先端をCに一致させる。水浴から取り出し、外部をよく拭いた後、質量W1を量る。同じピクノメーターで水を用いて同様に操作し、その規定温度t℃における質量W2を量る。第1法の式により比重dtt’を計算する。
③ 第3法 浮きばかりによる測定法
浮きばかりをエタノール又はエーテルで清浄にした後、試料をガラス棒でよくかき混ぜ、浮きばかりを入れ、規定温度にし、静止したとき、メニスカスの上線で比重を読む。読み方が表示してある浮きばかりでは、その方法に従う。
(31) ヒ素試験法
ヒ素試験法は、試料中に混在するヒ素の限度試験である。その限度は、三酸化ヒ素(As2O3)の量として表わす。
各条には、ヒ素(As2O3)の限度をμg/gで( )内に付記する。
装置A
図1に示すものを用いる。
排気管Bに、約30mmの高さまでガラス繊維Fを詰め、酢酸鉛試液及び水の等容量混液で均等に潤した後、下端から弱く吸引して過量の溶液を除く。これをゴム栓Hの中心に垂直に差し込み、Bの下部の小孔Eは、下に僅かに突き出るようにして発生瓶Aに付ける。Bの上端には、ガラス管Cを垂直に固定したゴム栓Jを付ける。Cの排気管側の下端は、ゴム栓Jと同一平面とする。
図1
| A:発生瓶(肩までの内容約70mL) |  |
| B:排気管 | |
| C:ガラス管(内径5.6mm,吸収管に入れる部分は先端を内径1mmに引き伸ばす。) | |
| D:吸収管(内径10mm) | |
| E:小孔 | |
| F:ガラス繊維(約0.2g) | |
| G:5mLの標線 | |
| H及びJ:ゴム栓 | |
| L:40mLの標線 |
装置B
図2に示すものを用いる。
排気管Bに、約30mmの高さまでガラス繊維Fを詰め、酢酸鉛試液及び水の等容量混液で均等に潤した後、下端から弱く吸引して過量の溶液を除く。これをゴム栓Hの中心に垂直に差し込み、Bの下部の小孔Eは、下に僅かに突き出るようにして発生瓶Aに付ける。Bの上端には、ガラス管Cを垂直に固定したゴム栓Jを付ける。Cの下端は、Jの下端と同一平面とする。次に、使用直前にC及びDのすり合わせ面の間に臭化第二水銀紙Gをはさみ、クリップKでC及びDを固定する。
図2
| A:発生瓶(肩までの内容約70mL) |  |
| B:排気管 | |
| C及びD:褐色ガラス管(内径5.5mm、接続部の外径18mmでそれぞれすり合わせとする。) | |
| E:小孔 | |
| F:ガラス繊維(約0.2g) | |
| G:臭化第二水銀紙(18mm×18mm) | |
| H及びJ:ゴム栓 | |
| K:クリップ | |
| L:40mLの標線 |
操作法
試料溶液の調製は、別に規定する場合を除き、次の方法によるものとする。
① 第1法
各条に規定する量の試料を量り、水5mLを加え、必要ならば、加温して溶かし、試料溶液とする。
② 第2法
各条に規定する量の試料を量り、水5mL及び硫酸1mLを加える。ただし、無機酸の場合には、硫酸を加えない。これに亜硫酸水10mLを加え、小ビーカーに入れ、水浴上で加熱して亜硫酸がなくなり約2mLとなるまで蒸発させ、水を加えて5mLとし、試料溶液とする。
③ 第3法
各条に規定する量の試料を量り、白金製、石英製又は磁製のるつぼにとる。これに硝酸マグネシウムのエタノール溶液(1→50)10mLを加え、エタノールに点火して燃焼させた後、徐々に加熱して灰化する。この方法で、なお炭化物が残るときは、少量の硝酸で潤し、再び強熱して灰化する。放冷した後、残留物に塩酸3mLを加え、水浴上で加温して溶かし、試料溶液とする。
試料溶液の試験は、別に規定する場合を除き、次の方法によるものとする。
① 装置Aを用いる方法
試料溶液を発生瓶Aに入れ、必要ならば、少量の水で洗い込む。これに、ブロムフェノールブルー試液1滴を加え、アンモニア試液、強アンモニア水又は希塩酸を用いて中和した後、塩酸(1→2)5mL及びヨウ化カリウム試液5mLを加え、2~3分間放置し、さらに、酸性塩化第一スズ試液5mLを加え、室温で10分間放置する。次に、水を加えて40mLとし、無ヒ素亜鉛2g(1.5~2.4g)を加え、30秒以内にB及びCを連結したゴム栓Hを発生瓶Aに付ける。Cの細管部の端は、あらかじめヒ化水素吸収液5mLを入れた吸収管Dの底に達するように入れておく。その後、発生瓶Aは、25℃の水中に肩まで浸し、1時間放置する。吸収管を外し、必要ならば、ピリジンを加えて5mLとし、吸収液の色を観察する。この場合において、吸収液の色は、標準色より濃くてはならない。なお、標準色の調製は、同時に行う。
② 装置Bを用いる方法
発生瓶Aに試料溶液を入れ、必要ならば、少量の水で洗い込む。これにメチルオレンジ試液1滴を加え、アンモニア試液、強アンモニア水又は希塩酸を用いて中和した後、塩酸(1→2)5mL及びヨウ化カリウム試液5mLを加え、2~3分間放置する。さらに、酸性塩化第一スズ試液5mLを加え、室温で10分間放置する。次に、水を加えて40mLとし、無ヒ素亜鉛2g(1.5~2.4g)を加え、30秒以内にB、C、D及びGを連結してゴム栓Hを発生瓶Aに付け、25℃の水中に発生瓶Aの肩まで浸し、1時間放置した後、30秒以内に臭化第二水銀紙の色を観察する。この色は、標準色より濃くてはならない。なお、標準色の調製は、同時に行う。
標準色の調製
標準色の調製は、別に規定する場合を除き、次の方法によるものとする。
発生瓶Aにヒ素標準液2mLを全量ピペットを用いて加え、さらに、塩酸(1→2)5mL及びヨウ化カリウム試液5mLを加え、2~3分間放置した後、酸性塩化第一スズ試液5mLを加え、室温で10分間放置する。以下試料溶液と同様に操作して得られた吸収液又は臭化第二水銀紙の呈色を標準色とする。この色は、三酸化ヒ素(As2O3)0.002mgに相当する。
標準液の調製
標準液の調製は、別に規定する場合を除き、次の方法によるものとする。
ヒ素標準原液:三酸化ヒ素を微細の粉末とし、105℃で4時間乾燥し、その100mg(99.5~100.4mg)を量り、水酸化ナトリウム溶液(1→5)5mLを加えて溶かす。この溶液に希硫酸を加えて中性とし、1,000mLの全量フラスコに入れ、更に希硫酸を追加し、新たに煮沸し冷却した水を標線まで加えて1,000mLとする。
ヒ素標準液:ヒ素標準原液10mLを全量ピペットを用いて量り、1,000mLの全量フラスコに入れ、希硫酸10mLを加え、新たに煮沸し冷却した水を標線まで加えて1,000mLとする。この溶液1mLは、三酸化ヒ素(As2O3)0.001mgを含む。この溶液は、用時調製し、共栓瓶に保存する。
注意:試験に用いる器具、試薬及び試液は、ヒ素を含まない又はほとんど含まないものを用い、必要ならば、空試験を行う。
(32) ビタミンA定量法
ビタミンA定量法は、ビタミンA油製造用原体、ビタミンA粉末製造用原体その他の飼料添加物中のビタミンAを紫外部の吸光度測定により定量する方法である。この場合において、定量を妨害する物質が存在するときは、適当な前処理を行う必要がある。
1ビタミンA単位(1ビタミンA国際単位と同じ。)は、ビタミンA(アルコール型)0.3μgに相当する。
試薬
イソプロパノール 水を対照液として、層長10mmで吸光度を測定するとき、波長300nmにおいて0.05以下、波長320~350nmにおいて0.01以下とする。必要ならば、蒸留して精製する。
エーテル 用時蒸留し、初めと終わりのそれぞれ約10%を除く。
操作法
遮光容器を用い、できる限り空気又は他の酸化剤との接触を避け、操作は、速やかに行う。
各条で別に規定する場合を除き、第1法を用いるが、第1法で測定できる条件に適合しないものには、第2法を用いる。
① 第1法
試料約0.5gを0.001gの桁まで量り、その数値を記録し、イソプロパノールを加えて溶かし、250mLの全量フラスコに入れ、更にイソプロパノールを標線まで加えて250mLとする。この溶液を、層長10mmで326nmにおける吸光度が約0.5となるように、イソプロパノールで正確に薄めて試料溶液とし、吸収極大の波長を測定する。また、層長10mmで300nm、310nm、320nm、326nm、330nm、340nm及び350nmにおける吸光度を測定し、326nmの吸光度を1.000としたときの各波長における吸光度の比を求める。吸収極大の波長が325~328nmの間にあり、かつ、得られた各波長における吸光度の比が、それぞれ表の値の±0.030の範囲内にあれば、326nmの吸光度Aから試料1g中のビタミンA単位を算出する。
1g中のビタミンA単位数=E1cm1%(326nm)×1,900
E1cm1%(326nm)=(A/W)×(V/100)
V:試料溶液の総mL数
W:試料溶液VmL中の試料のg数
酢酸レチノール及びパルミチン酸レチノールの確認のため、次の確認試験を行う。
試料、薄層クロマトグラフ用酢酸レチノール標準品及び薄層クロマトグラフ用パルミチン酸レチノール標準品についてそれぞれ15,000ビタミンA単位を含む量を量り、それぞれ石油エーテル5mLに溶かし、試料溶液及び標準液とする。この溶液につき、薄層クロマトグラフ法により試験を行う。試料溶液及び標準液5μLずつを薄層クロマトグラフ用シリカゲルを用いて調製した薄層板にスポットする。次に、ベンゼンを展開溶媒として約10cm展開した後、薄層板を風乾する。これに三塩化アンチモン試液を噴霧し、試料及び標準品の青色に呈色した主なスポットの位置を比較して確認する。
第1法により吸光度を測定し、吸収極大の波長が325~328nmの間にないとき又は吸光度の比が表示した値の±0.030の範囲内にないときは、第2法を用いる。
| λ(nm) | 酢酸レチノール | パルミチン酸レチノール |
| 300 310 320 326 330 340 350 |
0.578 0.815 0.948 1.000 0.972 0.786 0.523 |
0.590 0.825 0.950 1.000 0.981 0.795 0.527 |
② 第2法
別に規定する場合を除き、500ビタミンA単位以上に相当し、油脂1g以下を含む量の試料を有効数字3桁まで量り、その数値を記録し、フラスコに入れ、無アルデヒドエタノール30mL及びピロガロールのエタノール溶液(1→10)1mLを加える。次に、水酸化カリウム溶液(9→10)3mLを加え、還流冷却器を付け、水浴上で30分間加熱し、けん化する。速やかに常温まで冷却し、水30mLを加え、分液漏斗Aに移す。フラスコは、水10mLで洗った後エーテル40mLで洗い、洗液を分液漏斗Aに入れ、よく振り混ぜ、放置する。水層を分液漏斗Bに分取し、エーテル30mLでフラスコを洗った後、洗液を分液漏斗Bに入れ、振り混ぜ、抽出する。水層はフラスコに分取し、エーテル層は分液漏斗Aに合わせる。分取した水層は、分液漏斗Bに入れ、エーテル30mLを加え、振り混ぜ、抽出する。エーテル層は、分液漏斗Aに合わせる。これに水10mLを加え、静かに2~3回倒立した後静置し、分離した水層を除く。さらに、水50mLずつで3回洗い、回の進むにつれて次第に強く振る。洗液がフェノールフタレイン試液で呈色しなくなるまで、水50mLずつで洗った後、10分間放置する。水をできる限り除き、エーテル抽出液を三角フラスコに移し、エーテル10mLずつで2回洗い込む。次に、無水硫酸ナトリウム5g(4.5~5.4g)を加え、振り混ぜた後、傾斜してエーテル抽出液をナス型フラスコに移す。残った硫酸ナトリウムは、エーテル10mLずつで2回以上洗い、洗液をフラスコに合わせる。エーテル抽出液を45℃の水浴中で振り動かしながらアスピレーターを用いて濃縮して約1mLとし、30秒以内にイソプロパノールを加えて溶かし、1mL中に6~10ビタミンA単位を含むように正確に薄め、試料溶液とする。この溶液につき、層長10mmで波長310nm、325nm及び334nmにおける吸光度A1、A2及びA3を測定する。
1g中のビタミンA単位数=E1cm1%(325nm)×1,830
E1cm1%(325nm)=(A2/W)×(V/100)×f
f=6.815-2.555×(A1/A2)-4.260×(A3/A2)
f:補正係数
V:試料溶液の総mL数
W:試料溶液VmL中の試料のg数
(33) ビタミンD定量法
ビタミンD定量法は、ビタミンD3油製造用原体、ビタミンD粉末製造用原体その他の飼料添加物中のビタミンDを、ガスクロマトグラフ法により定量する方法である。ただし、この定量法は、ビタミンDに対するビタミンE(酢酸dl-α-トコフェロール)の質量比が2,500以下のものに適用される。
ビタミンD粉末製剤にあっては、8,000ビタミンD国際単位を含む量を有効数字3桁まで量り、その数値を記録し、アスコルビン酸ナトリウム溶液(1→20)20mLを加え、還流冷却器を付け、水浴中で泥状又は乳状とした後、これを試料として試験を行う。
ステロールが混在している場合にあっては、ケイソウ土・ジギトニンカラムによる脱ステリン操作を行う。試料をけん化抽出したベンゼン層50mLを減圧留去して得られた残留物に、n-ヘキサン3mLを加えて溶かしたものを、ケイソウ土・ジギトニンカラムに加えた後、n-ヘキサンを追加し、0.5mL/minの流速で流下させ、溶出液約30mLを集める。この溶出液の溶媒を留去した後、残留物にアセトン1.0mLを全量ピペットを用いて加え、溶かし、以下薄層クロマトグラフ用試料溶液として試験を行う。
1ビタミンD国際単位は、ビタミンD30.025μgに相当する。
試薬・試液
無アルデヒドエタノール エタノール〔特級〕1Lに50%水酸化カリウム溶液5mL及び亜鉛末5g(4.5~5.4g)を加え、約2時間還流した後、蒸留し、初めと終わりのそれぞれ約10%を除く。
n-ヘキサン〔特級〕 水を対照液とし、層長10mmで吸光度を測定するとき、波長240~250nmにおいて、E1cm1%=0.3以下のものを使用する。
ベンゼン ベンゼン〔特級〕を用時蒸留し、初めと終わりのそれぞれ約10%を除く。
アセトン アセトン〔特級〕に過マンガン酸カリウムを少量ずつ加え、振り混ぜ、2~3日放置して紫色が消えなくなった後、蒸留し、留液に新たに焼いた無水炭酸カリウムを加えて脱水し、分留管を付け、湿気を避けて蒸留し、56℃の留分を集める。
シリカゲル 薄層クロマトグラフ用(蛍光剤入り)
酢酸スチグマステロール スチグマステロール〔特級〕0.54g(0.535~0.544g)を量り、ピリジン4.8mLを加えて溶かし、無水酢酸1.2mLを加え、60~70℃の水浴中で1時間加温し、室温で一夜放置した後、これを水中に注ぎ込み、生じた沈殿をろ取する。この沈殿を水で洗った後、エタノールにより再結晶する(融点143~145℃)。
本法の試薬は、上記以外のものにあっては、日本産業規格試薬の特級の規格に適合するものを用いる。
標準品・標準液
ビタミンD2標準品 日本薬局方エルゴカルシフェロール この場合において、波長265nmにおける吸光度を測定するとき、E1cm1%=465(0.01g,エタノール,1,000mL)以上のものを用いる。
ビタミンD3標準品 日本薬局方コレカルシフェロール この場合において、波長265nmにおける吸光度を測定するとき、E1cm1%=470(0.01g,エタノール,1,000mL)以上のものを用いる。
ビタミンD・プレD溶液 ビタミンD標準品5mg(4.5~5.4mg)を量り、二塩化エチレン10mLを加えて溶かし、水浴中で30分間還流する。用時調製する。
内部標準液 A液 酢酸スチグマステロール0.050g(0.0495~0.0504g)を量り、アセトンを加えて溶かし、100mLの全量フラスコに入れ、更にアセトンを標線まで加えて100mLとする。冷暗所に保存する。
B液 A液10mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、アセトンを加えて溶かし、更にアセトンを標線まで加えて100mLとする。用時調製する。
ビタミンD標準液 ビタミンD0.040g(0.0395~0.0404g)を量り、アセトンを加えて溶かし、100mLの全量フラスコに入れ、更にアセトンを標線まで加えて100mLとする。この溶液10mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、内部標準液A液10mLを全量ピペットを用いて加え、アセトンを標線まで加えて100mLとし、ビタミンD標準液とする。用時調製する。
ケイソウ土・ジギトニンカラムの調製
ジギトニン600mg(599.5~600.4mg)に水10mLを加え、加温溶解し、約1時間放置した後、その5mLを量り、クロマトグラフ用ケイソウ土10g(9.5~10.4g)に加え、均等に混和し、その3g(2.5~3.4g)を量り、n-ヘキサンと共に10×300mmの褐色ガラス管に流し込み、大部分のn-ヘキサンを流出し、ケイソウ土・ジギトニンカラムを調製する。
操作法
遮光容器を用い、操作は、速やかに行う。
8,000ビタミンD国際単位を含む量の試料を有効数字3桁まで量り、その数値を記録し、フラスコに入れ、無アルデヒドエタノール50mL及びピロガロールのエタノール溶液(2→10)20mLを加える。次に、水酸化カリウム溶液(9→10)8mLを加え、還流冷却器を付け、水浴上で30分間加熱し、けん化する。速やかに常温まで冷却し、ベンゼン100mLを全量ピペットを用いて加え、栓をし、よく振り混ぜた後、分液漏斗に移し、これに水酸化カリウム試液40mLを加え、15秒間激しく振り混ぜた後、静置し、水層を除く。
ベンゼン層に水酸化カリウム溶液(3→100)40mLを加え、振り混ぜた後、静置し、水層を除く。これに水40mLを加え、静かに2~3回倒立した後、静置し、水層を除く。さらに、毎回水40mLずつで洗い、回の進むにつれて次第に強く振る。洗液がフェノールフタレイン試液で呈色しなくなるまで洗った後、水をできる限り除く。次に、乾燥した円形のろ紙(直径9cm)に切り込みを入れたものを加え、ベンゼン層が澄明になるまで振り混ぜる。
ベンゼン層50mLを全量ピペットを用いて量り、ガラス栓付100mLのナス型フラスコに入れ、40℃の水浴中で振り動かしながらアスピレーターを用いて減圧留去する。残留物にアセトン1.0mLを全量ピペットを用いて加え、栓をしてよく振り混ぜて溶かし、薄層クロマトグラフ用試料溶液とする。この試料溶液0.2mLを全量ピペット又はマイクロピペットを用いて量り、薄層クロマトグラフ用シリカゲル(蛍光剤入り)を用いて調製した薄層板にスポットする。別に、ビタミンD・プレD溶液を同一の薄層板のすみにスポットする。次に、n-ヘキサン・酢酸エチル混液(4:1)を展開溶媒として約15cm展開した後、薄層板を風乾し、紫外線(主波長254nm)を照射し、薄層クロマトグラフ用試料溶液から得たビタミンD及びプレDの部分をステンレス製ミクロスパーテルで5分以内にかきとり、50mLのビーカーに入れる。
アセトン5mLずつで6回抽出し、ろ紙を用いて50mLの丸底フラスコ中にろ過する。ろ紙は、少量のアセトンで洗い、洗液をろ液に合わせる。アセトン抽出液を、40℃の水浴中で振り動かしながらアスピレーターを用いて減圧留去する。速やかに室温に戻し、残留物に内部標準液B液0.50mLを全量ピペット又はマイクロピペットを用いて加え、溶かし、試料溶液とする。
試料溶液及びビタミンD標準液につき、次の条件でガスクロマトグラフ法により試験を行い、半値幅法によりピロD及び酢酸スチグマステロールそれぞれのピーク面積を求め、その面積比を求める。
試料1g中のビタミンDの国際単位数=S×(試料溶液の内部標準物質に対するピロDのピーク面積比/標準溶液の内部標準物質に対するピロDのピーク面積比)×V×(1/W)
S:標準液0.5mL中のビタミンDの国際単位数(標準液1mL中には、1,600国際単位のビタミンDを含む。)
V:希釈倍数(上記の場合は、2×5=10である。)
W:試料のg数
なお、操作条件は、次のとおりとする。
検出器:水素炎イオン化検出器
分離管:内径4mm、長さ1.5mのガラスカラム(1.5%メチルフェニルシリコーン-AW-DMCS 80~100メッシュ)
温度:分離管225℃ 試料注入口250℃ 検出器300℃
注入量:5μL
キャリヤーガス及び流速:窒素、内部標準物質が約40~60分後に現れるように窒素の流速を調整する。
(34) 沸点測定法及び蒸留試験法
沸点の測定及び蒸留試験は、別に規定する場合を除き、次の第1法又は第2法によるものとする。沸点は、最初の留液5滴が冷却器の先端から留出したときから、最後の留液がフラスコの底部から蒸発するときまでの温度とする。また、蒸留試験は、各条に規定する温度範囲の留分の容量を量るものである。
第1法 各条に規定する温度範囲が5℃未満のとき用いる。
装置
図に示すものを用いる。
| A:蒸留フラスコ |  |
| B:浸線付温度計 | |
| C:浸線 | |
| D:コルク栓 | |
| E:冷却器 | |
| F:アダプター | |
| G:メスシリンダー(25mL,0.1mLの目盛りのあるもの) |
操作法
あらかじめ液温を測定した試料25mLを0.1mLの目盛りのあるメスシリンダーGを用いて量り、内容50~60mLの蒸留フラスコAに入れ、このメスシリンダーを洗わずに受器とし、Aに沸騰石を入れ、浸線付温度計Bは、浸線Cがコルク栓Dの下端にくるように、また、水銀球の上端が留出口の中央部にくるように付け、Aに冷却器Eを連結し、EにはアダプターFを接続し、Fの先端は受器のメスシリンダーGの口に僅かに空気が流通するようにして差し込む。Aを覆う高さの風よけを付け、適当な熱源を用いてAを加熱する。
別に規定するものを除き、測定温度200℃未満のものは1分間4~5mL、200℃以上のものは1分間3~4mLの留出速度で蒸留し、留液の温度を初めの試料の液温と等しくし、留分の容量を量る。
80℃以下で蒸留し始める溶液では、試料をあらかじめ10~15℃に冷却し、その容量を量り、蒸留中は、メスシリンダーの上部から25mm以下を氷冷する。
気圧に対する温度の補正は、気圧100kPa未満のときは、0.36kPaにつき0.1℃を加え、気圧100kPaを超えるときは、0.36kPaにつき0.1℃を減じる。
第2法 各条に規定する温度範囲が5℃以上のとき用いる。
装置
第1法と同様の装置を用いる。ただし、蒸留フラスコAは、内容200mL、首の内径18~24mmで内径5~6mmの留出管が付いているものを用いる。
操作法
液温をあらかじめ測定した試料100mLを1mLの目盛りのあるメスシリンダーを用いて量り、第1法と同様に操作する。
(35) 融点測定法
融点とは、次のそれぞれの方法で測定した温度をいう。ある物質の融点が範囲で示されているときには、その物質の融点がその範囲内にあればよいことを示す。
その測定法は、飼料添加物の物理的・化学的性質により次の2方法に分ける。別に規定する場合を除き、第1法を用いる。
操作法
① 第1法
粉末にしやすいものは、この方法によるものとする。
試料を微細の粉末とし、別に規定する場合を除き、デシケーター(シリカゲル)で24時間乾燥する。また、各条に「乾燥した後」と規定されている場合は、乾燥減量の項の条件で乾燥したものを用いる。この試料を乾燥した毛細管Hに入れ、時計皿の上に立てた長さ約700mmのガラス管の内部に落とし、弾ませて固く詰め、層の厚さが2.5~3.5mmとなるようにする。
溶液Bを加熱し、予想した融点の約10℃下の温度まで徐々に上げ、浸線付温度計Dの浸線を溶液のメニスカスに合わせ、試料を入れた毛細管HをコイルスプリングGに挿入し、試料を詰めた部分がDの水銀球の中央にくるようにする。次に、1分間に約3℃上昇するように加熱して温度を上げ、予想した融点より約5℃低い温度から1分間に1℃上昇するように加熱を続ける。
試料がH内で液化して、固体を全く認めなくなったときのDの示度を読み取り、融点とする。
なお、装置は、図に示すものを用いることとし、溶液、浸線付温度計及び毛細管は、次のものを用いる。
溶液 常温における粘度50~100センチストークスの澄明なシリコーン油を用いる。
浸線付温度計 融点が50℃未満のときにあっては1号、50℃以上100℃未満のときにあっては2号、100℃以上150℃未満のときにあっては3号、150℃以上200℃未満のときにあっては4号、200℃以上250℃未満のときにあっては5号、250℃以上320℃未満のときにあっては6号を用いる。
毛細管 内径0.8~1.2mm、長さ120mm及び壁の厚さ0.2~0.3mmで一端を閉じた硬質ガラス製のものを用いる。
| A:加熱容器(硬質ガラス製) |  |
| B:溶液 | |
| C:テフロン製蓋 | |
| D:浸線付温度計 | |
| E:温度計固定ばね | |
| F:浴液量加減用小孔 | |
| G:コイルスプリング | |
| H:毛細管 | |
| J:テフロン製蓋固定ばね |
② 第2法
脂肪、脂肪酸、パラフィン又はろうのようなものであって、水に不溶で粉末にしにくいものは、この方法によるものとする。
注意しながら試料をできる限り低温で融解し、これを、泡が入らないようにして毛細管(第1法のもので両端を開いたもの)中に吸い上げ、約10mmの高さとする。毛細管から試料が流出しないように保ち、10℃以下で24時間放置し、又は少なくとも1時間氷上に放置した後、試料の位置が水銀球の中央外側にくるようにゴム輪で温度計に取り付け、水を入れたビーカーに入れ、試料の下端を水面下30mmの位置に保つ。水を絶えずかき混ぜながら加温し、予想した融点より5℃低い温度に達したとき、1分間に1℃上昇するように加熱を続ける。毛細管中で試料が浮上するときの温度を融点とする。
(36) 誘導結合プラズマ発光分光分析法及び誘導結合プラズマ質量分析法
誘導結合プラズマ発光分光分析法(以下「ICP発光分光分析法」という。)及び誘導結合プラズマ質量分析法(以下「ICP 質量分析法」という。)は、誘導結合プラズマ(Inductively Coupled Plasma)(以下「ICP」という。)を励起源又はイオン源として利用する元素分析法である。ICP発光分光分析法は、ICPにより励起された原子の原子発光スペクトル線の波長及び強度を測定する。ICP質量分析法は、検出器として質量分析計を用い、ICPによりイオン化された元素をm/z値ごとに分離してイオンのピーク強度を測定する。
装置
① ICP発光分光分析計の装置構成
ICP発光分光分析計は、励起源部、試料導入部、発光部、分光部、測光部及びデータ処理部で構成される。
励起源部は、発光部に電気エネルギーを供給・制御する ための高周波電源、制御回路及びガス供給部からなる。
試料導入部は、試料溶液を発光部に導入する部分で、試料溶液を霧化するネブライザー及び噴霧室(スプレーチャンバー)等から構成される。
発光部は、試料溶液中の元素を原子化・励起・発光させるための部分で、トーチ及び高周波誘導コイル等からなる。トーチは、三重管構造をしており、中心の管から試料溶液が導入される。プラズマの生成及び試料溶液を搬送するためのガスとしてアルゴンガスを用いる。発光部から放射される光の観測方式には、プラズマの側面の光を観測する横方向観測方式及びプラズマの中心の光を観測する軸方向観測方式がある。
分光部は、発光部から放射された光をスペクトル線に分離するための部分で、集光系及び回折格子等の光学素子からなる。分光器には、波長走査形分光器(モノクロメーター)と波長固定型の同時測定形分光器(ポリクロメーター)がある。なお、190nm以下の真空紫外領域のスペクトル線を測定する場合、分光器内は、真空排気を行うか、アルゴンガス又は窒素ガスにより、空気を置換する必要がある 。
測光部は、入射した光をその強度に応じた電気信号に変換する部分で、検出器及び信号処理系からなる。検出器としては、光電子増倍管又は半導体検出器が用いられる。
データ処理部は、データ処理を行い、検量線及び測定結果等を表示する。
② ICP質量分析計の装置構成
ICP質量分析計は、励起源部、試料導入部、イオン化部、インターフェース部、イオンレンズ部、質量分離部、イオン検出部及びデータ処理部で構成される。
励起源部、試料導入部及びイオン化部は、それぞれICP 発光分光分析計における励起源部、試料導入部及び発光部と同一の構造である。
インターフェース部は、大気圧下でプラズマにより生成されたイオンを高真空の質量分離部に導入するための境界部分でサンプリングコーン及びスキマーコーンより構成される。
イオンレンズ部は、インターフェース部を介して導入されたイオンを収束させ、効率良く質量分離部に導くための部分である。
質量分離部は、多くの装置で四重極型の質量分析計が採用されている。なお、コリジョン・リアクションセルと呼ばれる室(セル)を真空内の質量分離部の前に配置し、水素、ヘリウム、アンモニア又はメタン等のガスを導入することにより、後述の多原子イオン類による干渉を抑制できる。
イオン検出部は,検出器内に到達したイオンを、増倍管により増幅した後、電気信号に変換し、データ処理部で、 得られた電気信号をデータとして処理し、検量線及び測定結果等を表示する。
操作法
アルゴン又は窒素を所定の流量に設定し、高周波電源を入れ、プラズマを生成する。装置に指示された方法を用いて機器の校正を行う。別に規定する方法で調製した試料溶液、標準液又は比較液を導入し、ICP発光分光分析計の場合は適当な発光スペクトル線の発光強度を、ICP質量分析計の場合は定められたm/z値における信号強度を測定する。なお、定量に際しては、次の干渉及びバックグラウンドを考慮する必要がある。
① 操作条件の最適化
純度試験又は定量法を行うときは、あらかじめ次に規定する感度、バックグラウンド並びに酸化物イオン及び二価イオンの生成比の最適化を行い、装置の稼働性能が適切であることを確認しておく。操作条件の最適化の実施に際しては、通常、適切な濃度に調整した、7Li、9Be、59Co、89Y、115In、140Ce、205Tl、209Bi等の環境中から 汚染し難い、低質量数、中質量数及び高質量数を代表する元素の標準液を用いる。感度は、積分時間1秒当たりのイオンカウント数(以下「cps」という。)で判定する。純度試験又は定量法を行うときは、低質量数、中質量数及び 高質量数において、各元素濃度1μg/L(ppb)当たり数万cps程度あることが望ましい。
バックグラウンドは、天然には存在しない元素のm/z値、例えばm/zが4、8又は220等で測定した場合、10cps以下であることが望ましい。酸化物イオン及び二価イオンの生成比は、140Ce等の溶液を用い、それぞれの酸化物イオン(140Ceの場合140Ce16O+、m/z 156)、 二価イオン(140Ce2+、m/z 70)及び一価イオン(140Ce+、m/z 140)のカウント数を測定し、酸化物イオン及び二価イオンのカウント数を一価イオンのカウント数で除して求める。酸化イオン生成比、すなわち140Ce160+ /140Ce+が0.03以下及び二価イオン生成比、すなわち140Ce2+/140Ce+が0.05以下となることが望ましい。
② 干渉とその抑制又は補正
スペクトル干渉には、同重体干渉並びに多原子イオン及び二価イオンのマススペクトルの重なりによる干渉がある。同重体干渉とは、測定対象元素と原子量が近接している同重体イオンによる干渉をいう。例として、40Ca4に対する40Ar、204Pbに対する204Hgの重なりがある。多原子イオンは、イオン化源としてアルゴンガスを使用しているため、例えば、Arに起因する40Ar16O、40Ar16O1H、40Ar2等の多原子イオンが形成され、それぞれ56Fe、57Fe、80Seの測定に干渉を生じる。コリジョン・リアクションセルが付属している装置では、セル内でこれらの多原子イオンを減少させることができる。二価イオンとは、その一価イオンの1/2のm/z値にピークを持つイオンのことで、検液中に測定対象元素の2倍の質量数の同位体を持つ元素が共存する場合に干渉を生じる。非スペクトル干渉には、物理干渉及びイオン化干渉のほか、ICP質量分析法特有のものとしてマトリックス干渉がある。マトリックス干渉は多量の共存元素が存在すると測定対象元素のイオンカウント数が一般的に減少する現象である。この傾向は、共存元素の質量数が大きく、その濃度が高いほど、また、測定元素の質量数が小さいほど顕著に表れる。非スペクトル干渉は、未知試料に対して既知量の測定対象元素を添加することで、その回収率から干渉の程度を確認できる。回収率が低く、分析の信頼性が確保されないと判断される場合には、内標準法又は標準添加法によって補正を行う。
③ システムの再現性
各装置により最適化された試験条件の下、最低濃度の検量線用標準液を用いて、試験を6回繰り返すとき、別に規定するもののほか、分析対象元素のスペクトル強度の相対標準偏差は一定値以下(10%以下)であることを確認する。
定量は、通例、次のいずれかの方法による。
① 検量線法
3種以上の濃度の異なる標準液を調製し、それぞれの標準液につき、その強度を測定し、得られた値から検量線を作成する。次に測定可能な濃度範囲に調製した試料溶液の強度を測定した後、検量線から被検元素量(濃度)を求める。
② 標準添加法
同量の検液3個以上を量り、それぞれに被検元素が段階的に含まれるように標準液を添加し、更に溶媒を加えて一定容量とする。それぞれの溶液につき、強度を測定し、横軸に添加した標準被検元素量(濃度)、縦軸に強度をとり、グラフにそれぞれの値をプロットする。プロットから得られた回帰線を延長し、横軸との交点と原点との距離から被検元素量(濃度)を求める。ただし、この方法は、①による検量線が原点を通る直線の場合のみに適用できる。
③ 内標準法
内標準元素の一定量に対して標準被検元素を段階的に加えた標準液を数種類調製する。それぞれの液につき、各元素の分析線波長で標準被検元素による強度及び内標準元素による強度を同一条件で測定し、標準被検元素による強度と内標準元素による強度の比を求める。横軸に標準被検元素量(濃度)、縦軸に強度の比をとり、検量線を作成する。次に、標準液の場合と同量の内標準元素を加えた検液を調製し、検量線を作成したときと同一条件で得た被検元素による強度と内標準元素による強度の比を求め、検量線から被検元素量(濃度)を求める。なお、本法の適用に当たっては、添加する内標準元素が検液中に含まれないこと、又は含まれていたとしても添加濃度に対して無視できる程度であることを確認しておく必要がある。また、内標準元素としては、測定対象元素と、スペクトル干渉を起こさず、同程度のイオン化効率及び質量数を有する元素が望ましい。
注意
① アルゴンガスは、液化アルゴン又は圧縮アルゴンのいずれを用いても良いが、純度99.99vol%以上のものを用いる。
② 標準液の液性は検液と合わせることが望ましい。
③ 複数元素を含む標準液を調製する場合は、沈殿及び互いに干渉を生じないような試液並びに元素の組合せを選択する。
(37) 硫酸塩試験法
硫酸塩試験法は、試料中に混在する硫酸塩の限度試験である。
各条には、硫酸塩(SO4として)の限度を%で( )内に付記する。
操作法
別に規定する場合を除き、次の方法によるものとする。
各条に規定する量の試料をネスラー管に入れ、適量の水を加えて溶かし、40mLとする。これに希塩酸1mL及び水を加えて50mLとし、試料溶液とする。別に、各条で規定する量の0.005mol/L硫酸を量り、希塩酸1mL及び水を加えて50mLとし、比較液とする。この場合、試料溶液が澄明でないときは、両液を同条件でろ過する。
試料溶液及び比較液に塩化バリウム試液2mLずつを加え、混和し、10分間放置した後、黒色の背景を用い、ネスラー管の上方又は側方から観察して混濁を比較する。
試料溶液の呈する混濁は、比較液の呈する混濁より濃くてはならない。
(38) 硫酸呈色物試験法
硫酸呈色物試験法は、試料中に含まれる微量の不純物で硫酸により容易に着色する物質を試験する方法である。
操作法
あらかじめネスラー管を硫酸呈色物用硫酸でよく洗う。別に規定する場合を除き、試料が固体の場合には、ネスラー管に硫酸呈色物用硫酸5mLを入れ、試料を粉末とし、各条に規定する量を少量ずつ加え、ガラス棒でかき混ぜて完全に溶かす。試料が液体の場合には、各条に規定する量を量り、ネスラー管に入れ、硫酸呈色物用硫酸5mLを加え、振り混ぜる。この間、発熱し温度が上昇するものは冷却し、温度の影響のあるものは標準温度に保ち、15分間放置した後、溶液を白色の背景を用い、ネスラー管に入れた各条に規定する色の比較液と側方から観察して比色する。
(39) ろ紙クロマトグラフ法
ろ紙クロマトグラフ法は、ろ紙を用い、混合物を移動相で展開させてそれぞれの成分に分離する方法であり、物質の確認又は純度の試験等に用いる。
操作法
別に規定する場合を除き、次の方法によるものとする。
幅20~30mm、長さ400mmの長方形のろ紙の下端から約50mmの高さを原線とし、この中央に、各条で規定する量の試料溶液を、マイクロピペット又は毛細管を用いてスポットし、風乾する。次に、あらかじめ展開溶媒を入れ、その蒸気で飽和させておいた高さ約500mmの展開用容器に、このろ紙を入れ、器壁に触れないように注意して吊るし、下端から約10mmまでを器底の展開溶媒中に浸し、容器を密閉し、常温で展開を行う。
展開溶媒の先端が原線から各条に規定する距離まで上昇したとき、ろ紙を容器から取り出し、30秒以内に溶媒の先端の位置に印を付け、風乾した後、各条に規定する方法により、スポットの位置及び色等を調べる。Rf値は、次式により求める。
Rf=原線からスポットの中心までの距離/原線から溶媒先端までの距離
7 飼料添加物一般の試験法並びに各飼料添加物の成分規格及び製造方法等の基準に用いる標準品、試薬・試液、容量分析用標準液、標準液、色の比較液、計量器・用器、ろ紙、滅菌法及びベルトラン糖類定量表の規定
(1) 標準品
標準品は、一定の純度又は一定の生物学的作用を有するように調製された物質で、飼料添加物を生物学的又は理化学的に試験するときに用いるものである。
アスコルビン酸 C6H8O6〔日本薬局方標準品〕
L-アスコルビン酸-2-リン酸エステルトリスシクロヘキシルアンモニウム C6H6O9P・3[(CH2(CH2)4CHNH3]
含量 含量 本品は、定量するとき、L-アスコルビン酸-2-リン酸エステルトリスシクロヘキシルアンモニウム(C6H6O9P・3[(CH2(CH2)4CHNH3])98.0%以上を含む。
物理的・化学的性質 本品は、白色の粉末である。
確認試験 本品につき、赤外吸収スペクトル測定法の臭化カリウム錠剤法により測定するとき、波数2939cm-1、2859cm-1、1719cm-1、1586cm-1、1448cm-1、1389cm-1及び974cm-1付近に吸収を認める。
純度試験 本品0.01g(0.005~0.014g)を水20mLに溶かし、試料溶液とする。この溶液20μLにつき、次の条件で液体クロマトグラフ法の自動面積測定法によりピーク面積を測定し、面積百分率法によりL-アスコルビン酸-2-リン酸エステルトリスシクロヘキシルアンモニウム以外のピーク面積の和を求めるとき、2.0%以下である。
操作条件
検出器:紫外吸光光度計(測定波長:250nm)
カラム:内径4.6mm、長さ150mmのステンレス管に粒径5μmの液体クロマトグラフ用オクタデシルシリル化シリカゲルを充填する。
カラム温度:25℃付近の一定温度
移動相:リン酸二水素カリウム13.6g(13.55~13.64g)及びテトラブチルアンモニウムヒドロキシド試液4.0mLを水950mLに溶かし、2mol/L水酸化ナトリウム試液でpHを6.0に調整した後、水を加えて1,000mLとする。この溶液950mLにアセトニトリル50mLを加え、混和する。
流速:1.0mL/min
面積測定範囲:L-アスコルビン酸-2-リン酸エステルトリスシクロヘキシルアンモニウムの保持時間の6倍
乾燥減量 0.5%以下(0.1g,シリカゲル,24時間)
定量法 本品約11mgを0.1mgの桁まで量り、その数値を記録し、0.1mol/L塩酸を加えて溶かし、100mLの全量フラスコに入れ、0.1mol/L塩酸を標線まで加えて100mLとする。この溶液2.0mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、0.1mol/L塩酸・炭酸ナトリウム緩衝液(pH10)を標線まで加えて100mLとし、試料溶液とする。この試料溶液につき、0.1mol/L塩酸・炭酸ナトリウム緩衝液(pH10)を対照として263nm付近の極大波長における吸光度Aを測定する。
L-アスコルビン酸-2-リン酸エステルトリスシクロヘキシルアンモニウムの量(mg)=A/279.5×50,000
L-アスコルビン酸-2-リン酸エステルマグネシウム (C6H6O9P)2Mg3・10H2O
含量 本品は、定量するとき、98.0%以上を含む。
物理的・化学的性質 本品は、白色の粉末で、僅かに特異な臭いを有する。
確認試験
①① 本品2mg(1.5~2.4mg)に0.1mol/L塩酸試液を加えて100mLとし、この溶液につき、吸収スペクトルを測定するとき、波長235~239nmに吸収の極大を示す。
② 本品の水溶液(1→50)5mLに塩化第二鉄試液1滴を加えるとき、この溶液は、赤褐色を呈する。
③ 本品0.1g(0.05~0.14g)に硫酸2mL及び過酸化水素水15mLを加え、溶液が約5mLになるまで加熱し、放冷した後、水を加えて50mLとする。この溶液1mLに3mol/L硫酸1mL、モリブデン酸アンモニウム試液1mL及びアミドール・亜硫酸水素ナトリウム試液1mLを加えるとき、その溶液は、青色を呈する。
④ 本品0.5g(0.45~0.54g)に硫酸2mL及び過酸化水素水30mLを加え、溶液が約5mLになるまで加熱する。放冷した後、水を加えて20mLとし、1mol/L水酸化ナトリウム試液で中和した溶液は、マグネシウム塩の定性反応を呈する。
純度試験
① 溶状 本品1.0g(0.95~1.04g)に水10mLを加えて溶かすとき、その溶液は、色の比較液Jより濃くなく、澄明でなければならない。
② 類縁物質 本品0.01g(0.005~0.014g)に水1mLを加えて溶かし、この溶液10μLをクロマトグラフ用3号ろ紙の下端から約5cmのところにスポットし、風乾する。次に、トリクロル酢酸5g(4.5~5.4g)にイソプロパノール・水混液(75:20)95mLを加えて溶かした溶液を展開溶媒として約30cm展開した後、ろ紙を風乾する。これに塩化第二鉄0.5g(0.45~0.54g)をエタノールに溶かし、100mLとした溶液を均等に噴霧するとき、Rf値約0.5の位置に赤褐色の単一のスポットを認め、その他のスポットを認めてはならない。
水分 本品約1.6gを0.01gの桁まで量り、その数値を記録し、メタノール・硫酸混液(70:1)を加えて溶かした後、50mLの全量フラスコに入れ、標線まで加えて50mLとして試料溶液とし、質量を0.01gの桁まで量り、その数値を記録する。試料溶液1mLを全量ピペットを用いて量り、カールフィッシャー法の直接滴定法により水分を測定する。別に、0.01gの桁まで質量を量り、その数値を記録した約1mLのメタノール・硫酸混液(70:1)について同様に水分を測定する。次式により本品の水分を求めるとき、その量は、23.0~24.5%でなければならない。
本品の水分(%)=(50×S-B×(W2-W1)/W3)/(W1×10)
S:1mLの試料溶液中の水分(mg)
B:0.01gの桁まで質量を量り、その数値を記録した約1mLのメタノール・硫酸混液(70:1)中の水分(mg)
W1:本品の採取量(g)
W2:試料溶液50mLの質量(g)
W3:Bの測定に用いた約1mLのメタノール・硫酸混液(70:1)の質量(g)
定量法 本品0.2gを0.001gの桁まで量り、その数値を記録し、水を加えて溶かし、100mLの全量フラスコに入れ、更に水を標線まで加えて100mLとし、試料原液とする。この溶液25mLを全量ピペットを用いて量り、分解瓶に入れ、硫酸(1→3)1mL及びペルオキソ二硫酸カリウム(2→25)5mLを加え、密栓し、高圧蒸気滅菌器に入れ、121℃で30分間加熱する。放冷した後、100mLの全量フラスコに入れ、水を標線まで加えて100mLとする。この溶液2.5mLを全量ピペットを用いて量り、亜硫酸水素ナトリウム溶液(1→20)1mLを加え、振り混ぜ、さらに、モリブデン酸アンモニウム・タルトラトアンチモン(Ⅲ)酸カリウム・アスコルビン酸試液15mLを加え、振り混ぜた後、200mLの全量フラスコに入れ、水を標線まで加えて200mLとし、試料溶液とする。モリブデン酸アンモニウム・タルトラトアンチモン(Ⅲ)酸カリウム・アスコルビン酸試液を加えてから正確に30分後に、試料溶液につき、波長710nmにおける吸光度AT1を測定する。また、試料原液25mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、水を標線まで加えて100mLとし、この溶液2.5mLを全量ピペットを用いて量り、亜硫酸水素ナトリウム溶液(1→20)1mLを加え、振り混ぜ、以下試料溶液と同様に操作し、波長710nmにおける吸光度AT2を測定する。別に、リン酸二水素カリウム約0.1gを0.001gの桁まで量り、その数値を記録し、水を加えて溶かし、500mLの全量フラスコに入れ、更に水を標線まで加えて500mLとし、この溶液2.5mLを全量ピペットを用いて量り、亜硫酸水素ナトリウム溶液(1→20)1mLを加え、振り混ぜ、以下試料溶液と同様に操作し、波長710nmにおける吸光度ASを測定する。
L-アスコルビン酸-2-リン酸エステルマグネシウムの量(mg)=W×(4/5)×((AT1-AT2)/AS)×(379.61/136.09)×1,000
W:リン酸二水素カリウムの量(g)
4-アセタミド-2-ヒドロキシ安息香酸メチル C10H11NO4
含量 本品は、105℃で2時間乾燥した後、定量するとき、4-アセタミド-2-ヒドロキシ安息香酸メチル(C10H11NO4)98.0%以上を含む。
物理的・化学的性質 本品は、白色の結晶性の粉末で、臭いはない。
確認試験
① 本品のメタノール溶液(1→125,000)につき、吸収スペクトルを測定するとき、波長211~215nm、272~276nm及び305~309nmに吸収の極大を示し、波長237~241nm及び291~295nmに吸収の極小を示す。
② 本品を105℃で2時間乾燥し、赤外吸収スペクトル測定法の臭化カリウム錠剤法により赤外吸収スペクトルを測定するとき、3,300cm-1、1,680cm-1、1,615cm-1、1,320cm-1及び1,275cm-1に吸収を認める。
純度試験
① 融点 本品の融点は、151~153℃でなければならない。
② 類縁物質 本品0.01g(0.005~0.014g)を量り、メタノール1.0mLを加えて溶かし、この溶液10μLを、薄層クロマトグラフ用シリカゲルを用いて調製した薄層板にスポットする。次に、エーテル・ベンゼン・クロロホルム混液(50:35:15)を展開溶媒として約10cm展開した後、薄層板を風乾する。これにドラーゲンドルフ試液を均等に噴霧し、その後、硫酸(1→2)を均等に噴霧するとき、Rf値約0.3の位置に橙色の単一のスポットを認め、その他のスポットを認めてはならない。
乾燥減量 1.0%以下(1g,105℃,2時間)
強熱残分 0.10%以下(1g)
定量法 本品を105℃で2時間乾燥し、その約0.045gを0.0001gの桁まで量り、その数値を記録し、窒素定量法により試験を行う。
0.01mol/L硫酸1mL=4.184mgC10H11NO4
注意:遮光した気密容器に保存する。
4-アミノ-2-エトキシ安息香酸メチル C10H13NO3
含量 本品は、デシケーター(シリカゲル)で3時間乾燥した後、定量するとき、4-アミノ-2-エトキシ安息香酸メチル(C10H13NO3)98.0%以上を含む。
物理的・化学的性質
① 本品は、灰褐色の結晶性の粉末で、臭いはない。
② 融点 99~101℃(分解)
確認試験
① 本品のメタノール溶液(1→125,000)につき、吸収スペクトルを測定するとき、波長208~212nm、232~236nm、277~281nm及び300~304nmに吸収の極大を示し、波長222~226nm、250~254nm及び287~291nmに吸収の極小を示す。
② 本品をデシケーター(シリカゲル)で3時間乾燥し、赤外吸収スペクトル測定法の臭化カリウム錠剤法により赤外吸収スペクトルを測定するとき、3,400cm-1、1,695cm-1、1,610cm-1及び1,255cm-1に吸収を認める。
純度試験 類縁物質 本品0.010g(0.0095~0.0104g)を量り、メタノール1.0mLを加えて溶かし、この溶液10μLを薄層クロマトグラフ用シリカゲルを用いて調製した薄層板にスポットする。次に、エーテル・ベンゼン・クロロホルム混液(50:35:15)を展開溶媒として約10cm展開した後、薄層板を風乾する。これにドラーゲンドルフ試液を均等に噴霧し、その後、硫酸(1→2)を均等に噴霧するとき、Rf値約0.5の位置に橙色の単一のスポットを認め、その他のスポットを認めてはならない。
乾燥減量 3.0%以下(1g,シリカゲル,3時間)
強熱残分 0.10%以下(1g)
定量法 本品をデシケーター(シリカゲル)で3時間乾燥し、その約0.04gを0.0001gの桁まで量り、その数値を記録し、窒素定量法により試験を行う。
0.01mol/L硫酸1mL=3.904mgC10H13NO3
注意:遮光した気密容器に保存する。
アンプロリウム C14H19ClN4・HCl 本品は、アンプロリウム製造用原体を量り、氷酢酸を用いて再結晶し、アセトンで洗浄した後、水に加温溶解させ、イソプロパノールを加えて再び再結晶させ、アセトンで洗浄して調製する。
物理的・化学的性質
① 本品は、白色の結晶性の粉末で、臭いはない。
② 融点 約248℃(分解)
確認試験
① 本品の0.1mol/L塩酸溶液(1→100,000)につき、吸収スペクトルを測定するとき、波長244~248nm及び260~264nmに吸収の極大を示し、それぞれの極大波長における吸光度をA1及びA2とするとき、A1/A2は、1.04~1.06である。
② 本品の水溶液(1→50)は、塩化物の定性反応を呈する。
純度試験 類縁物質 本品0.10g(0.095~0.104g)を量り、メタノール10mLを加えて溶かし、この溶液10μLを、薄層クロマトグラフ用シリカゲルを用いて調製した薄層板にスポットする。次に、水・第二ブタノール・氷酢酸混液(5:4:1)を展開溶媒として約10cm展開した後、薄層板を風乾する。これにドラーゲンドルフ試液を均等に噴霧し、その後、硫酸(1→2)を均等に噴霧するとき、Rf値約0.4の位置に橙色の単一のスポットを認め、その他のスポットを認めてはならない。
乾燥減量 0.5%以下(1g,減圧,100℃,3時間)
強熱残分 0.10%以下(1g)
注意:遮光した気密容器に保存する。
エトパベート C12H15NO4 本品は、エトパベート製造用原体を量り、メタノールを用いて再結晶して調製する。
物理的・化学的性質 本品は、白色~僅かに微黄白色の結晶性の粉末で、臭いはない。
確認試験 本品のメタノール溶液(1→125,000)につき、吸収スペクトルを測定するとき、波長266~270nm及び297~301nmに吸収の極大を示し、波長236~240nm及び285~289nmに吸収の極小を示す。
純度試験
① 融点 本品の融点は、147~151℃でなければならない。
② 類縁物質 本品0.010g(0.0095~0.0104g)を量り、メタノール1.0mLを加えて溶かし、この溶液10μLを、薄層クロマトグラフ用シリカゲルを用いて調製した薄層板にスポットする。次に、クロロホルム・エーテル・メタノール混液(10:9:1)を展開溶媒として約10cm展開した後、薄層板を風乾する。これを亜硝酸ナトリウム10g(9.5~10.4g)に塩酸20mLを加えて亜硝酸ガスを発生させた容器中で10分間放置した後、これにクロモトロプ酸0.05g(0.045~0.054g)及び酢酸ナトリウム40g(39.5~40.4g)を水に溶かし、100mLとした溶液を均等に噴霧するとき、Rf値約0.5の位置に赤紫色の単一のスポットを認め、その他のスポットを認めてはならない。
乾燥減量 0.5%以下(1g,減圧,100℃,2時間)
強熱残分 0.10%以下(1g)
注意:遮光した気密容器に保存する。
塩酸チアミン C12H17ClN4OS・HCl〔日本薬局方標準品〕
塩酸ピリドキシン C8H11NO3・HCl〔日本薬局方標準品〕
クエン酸モランテル C12H16N2S・C6H8O7・H2O 本品は、クエン酸モランテル製造用原体を量り、光を避けて水で2回再結晶して調製する。
物理的・化学的性質
① 本品は、淡黄色の結晶性の粉末で、味は僅かに苦く、特異な臭いを有する。
② 本品は、メタノールにやや溶けやすく、水又はエタノールに溶けにくく、酢酸エチル又はベンゼンにほとんど溶けない。
③ 本品の水溶液(1→200)のpHは、3.5~4.3である。
④ 融点 117~120℃
確認試験
① 本品0.1g(0.05~0.14g)に水30mLを加えて溶かす。この溶液0.5mLにp -ジメチルアミノベンズアルデヒド・塩化第二鉄試液3mLを加えるとき、溶液は、赤紫色を呈する。
② 本品0.01g(0.005~0.014g)に水2mLを加えて溶かし、過マンガン酸カリウム試液1滴を加えるとき、試液の色は、30秒以内に消える。
③ 本品5mg(4.5~5.4mg)にクエン酸の無水酢酸溶液(0.5→100)2mLを加え、水浴中で加熱するとき、赤色~赤紫色を呈する。
④ 本品0.01g(0.005~0.014g)に0.01mol/L塩酸・メタノール試液を加えて溶かし、1,000mLとする。この溶液につき、吸収スペクトルを測定するとき、波長322~327nmに吸収の極大を示す。
⑤ 本品0.02g(0.015~0.024g)に水4mLを加えて溶かし、希水酸化ナトリウム試液を加えて中性とした溶液は、クエン酸塩の定性反応③を呈する。
純度試験
① 溶状 本品0.5g(0.45~0.54g)にメタノール10mLを加えて溶かすとき、その溶液は、黄色で、澄明でなければならない。
② シス異性体 本品0.2g(0.15~0.24g)をメタノールを加えて溶かし、10mLの褐色全量フラスコに入れ、更にメタノールを標線まで加えて10mLとし、試料溶液とする。この溶液1mLを全量ピペットを用いて量り、10mLの褐色全量フラスコに入れ、メタノールを標線まで加えて100mLとし、シス異性体用対照溶液とする。別に、無水クエン酸0.1g(0.05~0.14g)を量り、メタノールを加えて溶かし、100mLの全量フラスコに入れ、更にメタノールを標線まで加えて100mLとし、クエン酸溶液とする。試料溶液、シス異性体用対照溶液及びクエン酸溶液それぞれ5μLずつを薄層クロマトグラフ用シリカゲルを用いて調製した薄層板に暗所でスポットする。次に、メチルイソブチルケトン・ギ酸・水混液(2:1:1)の上層を展開溶媒として暗所で約10cm展開した後、薄層板を100℃で15分間乾燥する。この薄層板をヨウ素蒸気を満たした槽中に入れるとき、試料溶液から得たモランテル及びクエン酸以外のスポットは、認めない、又はシス異性体用対照溶液から得たスポットより濃くてはならない(1%以下)。
水分 3.7~4.7%(0.5g)
強熱残分 0.15%以下(1.0g)
酢酸レチノール、薄層クロマトグラフ用 C22H32O2〔薄層クロマトグラフ用酢酸レチノール、日本薬局方標準品〕
シアノコバラミン C63H88CoN14O14P〔日本薬局方標準品〕
スルファキノキサリン C14H12N4O2S 本品は、スルファキノキサリン製造用原体を量り、エタノールを用いて再結晶して調製する。
物理的・化学的性質
① 本品は、淡黄色の微細な結晶で、臭いはない。
② 本品は、アセトンに溶けにくく、エタノールに極めて溶けにくく、水にほとんど溶けない。
③ 本品は、1mol/L水酸化ナトリウム試液又は炭酸ナトリウム試液に溶ける。
④ 本品は、光により徐々に暗色となる。
確認試験 本品0.02g(0.015~0.024g)を量り、水5mLを加え、かき混ぜながら1mol/L水酸化ナトリウム試液を滴加して溶かし、これに硫酸銅試液2~3滴を加えるとき、黄緑色の沈殿を生じる。
純度試験
① 融点 本品の融点は、245~247℃(分解)でなければならない。
② 類縁物質 本品0.10g(0.095~0.104g)を量り、アセトン20mLを加え、加温して溶かし、この溶液20μLを、薄層クロマトグラフ用シリカゲルを用いて調製した薄層板にスポットする。次に、イソプロパノール・酢酸ブチル・水・強アンモニア水混液(10:6:3:1)を展開溶媒として約10cm展開した後、薄層板を風乾する。これを亜硝酸ナトリウム10g(9.5~10.4g)に塩酸20mLを加えて亜硝酸ガスを発生させた容器中で10分間放置した後、これにクロモトロプ酸0.05g(0.045~0.054g)及び酢酸ナトリウム40g(39.5~40.4g)を水に溶かし、100mLとした溶液を均等に噴霧するとき、Rf値約0.5の位置に紫赤色の単一のスポットを認め、その他のスポットを認めてはならない。
乾燥減量 0.5%以下(1g,105℃,4時間)
強熱残分 0.10%以下(1g)
注意:遮光した気密容器に保存する。
チロシン C9H11NO3〔日本薬局方標準品〕
ナイカルバジン C13H10N4O5・C6H8N2O 本品は、ナイカルバジン製造用原体を量り、ジオキサン・アセトン・水混液(5:4:1)を用いて再結晶して調製する。
物理的・化学的性質
① 本品は、帯黄色の粉末で、臭いはない、又は僅かに特異な臭いを有する。
② 融点 約260℃(分解)
確認試験
① 本品の無水エタノール溶液(1→15,000)15mLにスルファニル酸試液(2→125)5mL及び新たに調製した亜硝酸ナトリウム溶液(1→100)5mLを加え、密栓し、65℃で10分間加温するとき、溶液は、赤色を呈する。
② 本品の無水エタノール溶液(1→15,000)15mLに水酸化カリウムのエタノール溶液(1→100)5mLを加えるとき、溶液は、黄色を呈する。
純度試験 類縁物質 本品10mg(9.5~10.4mg)を量り、ジメチルホルムアミド1.0mLを加えて溶かし、この溶液5μLを、薄層クロマトグラフ用セルロース(蛍光剤入り)を用いて調製した薄層板にスポットする。次に、クロロホルム・エーテル・メタノール(10:9:1)を展開溶媒として約10cm展開した後、薄層板を風乾する。これに紫外線(主波長254nm)を照射するとき、Rf値約0.3の位置に単一のスポットを認め、その他のスポットを認めてはならない。
乾燥減量 0.5%以下(1g,減圧,五酸化リン,110℃,1時間)
強熱残分 0.10%以下(1g)
注意:遮光した気密容器に保存する。
パラアミノベンゾイルグルタミン酸 C11H14N2O3〔日本薬局方標準品〕
パルミチン酸レチノール、薄層クロマトグラフ用 C36H60O2〔薄層クロマトグラフ用パルミチン酸レチノール、日本薬局方標準品〕
葉酸 C19H19N7O6〔日本薬局方標準品〕
リボフラビン C17H20N4O6〔日本薬局方標準品〕
(2) 試薬・試液
試薬は、飼料添加物の試験に用いるものである。「容量分析用標準試薬」、「特級」、「1級」又は「無ヒ素」等と記載したものは、それぞれ日本産業規格試薬の容量分析用標準試薬、特級、1級又は無ヒ素等の規格に適合するものであり、かつ、試験法は、日本産業規格試薬の試験法に従う。試薬名が日本産業規格と相違する場合は、これを併記する。「日局」と記載したものは、日本薬局方の医薬品各条の成分規格に適合するものである。単に試験法を記載してある試薬については、飼料添加物の試験法を準用する。
試液は、飼料添加物の試験に用いるために調製した溶液である。
亜鉛(標準試薬) Zn〔容量分析用標準試薬〕
亜鉛、無ヒ素 Zn〔無ヒ素〕 約800μmのものを用いる。
亜鉛末 Zn〔特級〕
亜硝酸ナトリウム NaNO2〔特級〕
亜硝酸ナトリウム試液 亜硝酸ナトリウム10g(9.5~10.4g)に水を加えて溶かし、100mLとする。用時調製する。
アスコルビン酸 C6H8O6〔日局〕
アスコルビン酸ナトリウム C6H7NaO6〔特級〕
アセチレン C2H2〔溶解性アセチレン〕98.0%以上
アセトン CH3COCH3〔特級〕
ビタミンD定量に用いるアセトンは、一般試験法のビタミンD定量法に定めるところによるものとする。
アセトン、非水滴定用 アセトンに過マンガン酸カリウムを少量ずつ加え、振り混ぜ、2~3日放置して紫色が消えなくなった後、蒸留し、留液に新たに焼いた無水炭酸カリウムを加えて脱水し、分留管を付け、湿気を避けて蒸留し、56℃の留分を集める。
アセトニトリル CH3CN〔特級〕
アセトニトリル、液体クロマトグラフ用 CH3CN 無色澄明の液で水と混和する。水を対照液として、層長10mmで吸光度を測定するとき、波長200nmにおいて0.07以下、波長210nmにおいて0.046以下、波長220nmにおいて0.027以下、波長230nmにおいて0.014以下、波長240nmにおいて0.009以下のものとする。
アニリン C6H5NH2〔特級〕
アミグダリン C20H27NO11 本品は、水にやや溶けやすく、エタノールに溶けにくく、エーテルにほとんど溶けない。
pH 本品の水溶液(1→100)のpHは、4.5~6.5でなければならない。
融点 210~222℃
比旋光度 〔α〕D20=-39~-43°
乾燥減量 5%以下
強熱残分 0.1%以下
アミドール (NH2)2C6H3OH・2HCl 微黄褐色~灰黄緑色の結晶性の粉末である。
溶状 本品1.0g(0.95~1.04g)に水20mLを加えて溶かすとき、溶液は、澄明である。
含量 98.0%以上
アミドール・亜硫酸水素ナトリウム試液 アミドール0.4g(0.35~0.44g)及び亜硫酸水素ナトリウム8g(7.5~8.4g)を量り、水を加えて溶かし、40mLとする。用時調製する。
4-アミノアンチピリン C11H13N3O〔特級〕
アミノ酸分析用ニンヒドリン ニンヒドリン、アミノ酸分析用の項に定める。
アミノ酸分析用ニンヒドリン試液 ニンヒドリン試液、アミノ酸分析用の項に定める。
アミノピリン C13H17N3O 無色若しくは白色の結晶又は白色の結晶性粉末で、臭いはなく、味は僅かに苦い。
融点 107~109℃
乾燥減量 0.5%以下(1g,シリカゲル,4時間)
強熱残分 0.10%以下(1g)
遮光して保存する。
p-アミノ安息香酸 NH2C6H4COOH〔特級〕
アミルアルコール、イソ (CH3)2CH2CH2CHOH〔特級〕
アラビノース C5H10O5
比旋光度 〔α〕D20=-103~-105°
重金属 10μg/g以下
水分 0.5%以下
アリザリンエローGG C13H8N3NaO5〔特級〕 変色範囲 pH(黄色)10.0~12.0(褐色)
アリザリンエローGG試液 アリザリンエローGG0.1g(0.05~0.14g)にエタノール100mLを加えて溶かし、必要ならば、ろ過する。
アリザリンエローGG・チモールフタレイン試液 アリザリンエローGG試液10mLにチモールフタレイン試液20mLを加え、混和する。
アリザリンスルホン酸ナトリウム C14H5O2(OH)2SO3Na・H2O〔特級〕
アリザリンレッドS C14H5O2(OH)2SO3Na・H2O〔アリザリンレッドS(アリザリンスルホン酸ナトリウム)、特級〕 変色範囲pH(黄)3.7~5.2(橙赤)
アリザリンレッドS試液 アリザリンレッドS0.1g(0.05~0.14g)に水を加えて溶かし、100mLとする。必要ならば、ろ過する。
亜硫酸水素ナトリウム NaHSO3〔特級〕
亜硫酸ナトリウム Na2SO3・7H2O〔特級〕
アルカリ性銅試液A リン酸一水素ナトリウム71g(70.5~71.4g)及び酒石酸カリウムナトリウム40g(39.5~40.4g)を水650mLに溶かし、1mol/L水酸化ナトリウム試液100mLを加え、これを静かにかき混ぜながら、さらに、硫酸銅溶液(10→100)80mLを徐々に加える。次に、無水硫酸ナトリウム180g(179.5~180.4g)を加えて溶かした後、ヨウ素酸カリウム溶液(3.6→100)25mLを加え、さらに、水を加えて1,000mLとする。2日間25~35℃で放置した後、沈殿物をろ過して除き、25~35℃で保存する。
アルカリ性銅試液B 硫酸銅4.0g(3.95~4.04g)、無水炭酸ナトリウム24g(23.5~24.4g)、炭酸水素ナトリウム16g(15.5~16.4g)、無水硫酸ナトリウム180g(179.5~180.4g)及び酒石酸カリウムナトリウム12g(11.5~12.4g)に水を加えて溶かし、900mLとする。この溶液を10分間沸騰させた後、冷却し、水を加えて1,000mLとし、密栓し、1週間放置した後、ガラスろ過器(G3)でろ過し、遮光して保存する。
アルカリ性ブルーテトラゾリウム試液 ブルーテトラゾリウム試液、アルカリ性の項に定める。
アルミニウム Al〔特級〕
安息香酸(標準試薬) C6H5COOH〔容量分析用標準試薬〕
安息香酸プロピル C6H5COOCH2CH2CH3 無色澄明の液体である。
含量 98.0%以上
安息香酸プロピル・ジメチルホルムアミド試液 安息香酸プロピル1.0g(0.95~1.04g)にジメチルホルムアミドを加えて溶かし、100mLとする。
安息香酸ベンジル C6H5COOCH2C6H5〔日局〕
アントロン C14H10O〔特級〕
アントロン試液 アントロン35mg(34.5~35.4mg)に硫酸100mLを加えて溶かす。
アンモニア・塩化アンモニウム緩衝液、pH10.7 塩化アンモニウム67.5g(67.45~67.54g)に水を加えて溶かし、強アンモニア水570mLを加え、次に、水を加えて1,000mLとする。
アンモニア試液 強アンモニア水400mLに水を加えて1,000mLとする(10%)。
アンモニア水、強 NH4OH〔アンモニア水、特級、比重0.90〕
アンモニア水、25% NH4OH〔アンモニア水、特級、比重0.91〕
アンモニア水 〔日局〕
アンモニア性メタノール試液 強アンモニア水10mLにメタノール190mLを加える。
イソアミルアルコール アミルアルコール、イソの項に定める。
イソオクタン (CH3)3CCH2CH(CH3)2〔日局〕
イソブタノール ブタノール、イソの項に定める。
イソプロパノール プロパノール、イソの項に定める。
L-イソロイシン、定量用 乾燥したものを定量するとき、L-イソロイシン(C6H13NO2)99.0%以上を含むもの。
一酸化鉛 PbO〔特級〕
牛血清アルブミン 牛の血清から分離し、アルコール分画法で精製されたアルブミンの白色~薄い黄褐色の粉末である。
純度 96%以上
牛心臓抽出液 本品は、脂肪、腱及び血管を除いた心筋を肉ひき機で細挫し、水を加え、ときどき振りながら24時間4℃以下で保存する。次に、50℃の水浴中で数時間加温した後、沸騰させ、又は蒸気下で100℃に数分間放置する。放冷した後、布で、次に、ろ紙でろ過する。
馬脱繊維血液 無菌的に採血後30秒以内に滅菌ガラス玉で確実に脱繊維する。
保存法 2~6℃
使用期限 製造後2週間
ウラシル C4H4N2O2〔1級〕
液体クロマトグラフ用アセトニトリル アセトニトリル、液体クロマトグラフ用に定める。
液体クロマトグラフ用オクタデシルシリル化シリカゲル〔日局〕
液体クロマトグラフ用シリカゲル シリカゲル、液体クロマトグラフ用の項に定める。
エステル分解酵素液 本品は、Streptomyces rochei var. volubilis の培養ろ液から分離したエステル分解酵素を硫酸アンモニウム溶液(423→1,000)に懸濁した溶液であり、1.0単位/mL以上を含む。なお、1単位は、25℃、pH7.0で1分間に酢酸4-ニトロフェニルから4-ニトロフェノール1μmolを生成する量である。本品は、6℃以下に保存する。本品は、用時、適量の水で薄め、メンブランフィルター(0.45μm)でろ過した後、更に水で薄めて0.035単位/mLのエステル分解酵素液とする。
エタノール C2H5OH〔エチルアルコール(95v/v%)、特級〕
エタノール(99.5) エタノール、無水の項に定める。
エタノール、希 エタノール1容量に水1容量を加える。C2H5OH47.45~50.00v/v%を含む。
エタノール、中和 エタノール適量にフェノールフタレイン試液2~3滴を加え、これに0.01mol/L水酸化ナトリウム溶液又は0.1mol/L水酸化ナトリウム溶液を、液が淡赤色を呈するまで加える。用時調製する。
エタノール、不含クロロホルム クロロホルム、エタノール不含の項に定める。
エタノール、無アルデヒド エタノール1Lを共栓瓶に入れ、酢酸鉛2.5g(2.45~2.54g)を水5mLに溶かした溶液を加え、よく混ぜる。別に、水酸化カリウム5g(4.5~5.4g)を温エタノール25mLに溶かし、放冷した後、この溶液を前の溶液にかき混ぜないで静かに加え、1時間後この溶液を激しく振り混ぜ、一夜放置し、上澄液をとり、蒸留する。
ビタミンD定量に用いるものは、一般試験法のビタミンD定量法に定めるところによるものとする。
エタノール、無水 C2H5OH〔エチルアルコール(99.5v/v%以上)、特級〕
エチレングリコール HOCH2CH2OH〔エチレングリコール(グリコール)、特級〕
エチレングリコール、カールフィッシャー用 エチレングリコールを蒸留し、195~198℃の留分をとる。本品1mL中の水分は、1.0mg以下である。
エチレングリコールモノメチルエーテル HOCH2CH2OCH3〔エチレングリコールモノメチルエーテル(メチルセロソルブ)、特級〕
エチレンジアミン四酢酸二ナトリウム C10H14N2Na2O8・2H2O〔特級〕
エーテル C2H5OC2H5〔エチルエーテル、特級〕
エトキシキン、定量用 エトキシキン製造用原体 定量するとき、エトキシキン(C14H19NO)98.0%以上のものに限る。
NN指示薬 2-オキシ-1-(2′-オキシ-4′-スルホ-1′-ナフチルアゾ)-3-ナフトエ酸0.5g(0.45~0.54g)及び無水硫酸ナトリウム50g(49.5~50.4g)を混ぜ、均質になるまですりつぶして調製する。
エピクロルヒドリン C3H5OCl〔特級〕
エリオクロムブラックT C20H12N3NaO7S〔エリオクロムブラックT、(1-(1-ハイドロオキシ-2-ナフチルアゾ)-5-ニトロ-2-ナフトール-4-スルホン酸ナトリウム)、特級〕
エリオクロムブラックT・塩化ナトリウム指示薬 エリオクロムブラックT0.1g(0.05~0.14g)及び塩化ナトリウム10g(9.5~10.4g)を混ぜ、均質になるまですりつぶして調製する。
エリオクロムブラックT試液 エリオクロムブラックT0.5g(0.45~0.54g)及び塩酸ヒドロキシルアミン4.5g(4.45~4.54g)を量り、エタノール100mLを加えて溶かす。遮光した容器に保存する。
塩化アンモニウム NH4Cl〔特級〕
塩化アンモニウム試液 塩化アンモニウム10.5g(10.45~10.54g)に水を加えて溶かし、100mLとする(2mol/L)。
二塩化エチレン 一般試験法のビタミンD定量法に定めるところによる。
塩化カリウム KCl〔特級〕
塩化カリウム・塩酸緩衝液 塩化カリウム溶液(3→20)250mL及び2mol/L塩酸試液53mLに水を加えて1,000mLとする。
塩化カルシウム CaCl2・2H2O〔塩化カルシウム(2水塩)、特級〕
塩化カルシウム、水分測定用 CaCl2〔水分測定用、1~3号〕
塩化カルシウム試液 塩化カルシウム7.5g(7.45~7.54g)に水を加えて溶かし、100mLとする(0.5mol/L)。
塩化第一スズ SnCl2・2H2O〔特級〕
塩化第一スズ試液、酸性 塩化第一スズ8g(7.5~8.4g)に塩酸500mLを加えて溶かす。共栓瓶に保存する。調製した後3か月以内に用いる。
塩化第二水銀 HgCl2〔特級〕
塩化第二水銀試液 塩化第二水銀6.5g(6.45~6.54g)に水を加えて溶かし、100mLとする(0.25mol/L)。
塩化第二鉄 FeCl3・6H2O〔特級〕
塩化第二鉄試液 塩化第二鉄9g(8.5~9.4g)に水を加えて溶かし、100mLとする(0.5mol/L)。
塩化第二鉄試液、希 塩化第二鉄試液2mLに水を加えて100mLとする。用時調製する。
塩化第二鉄・塩酸試液 塩化第二鉄10g(9.5~10.4g)に0.1mol/L塩酸を加えて溶かし、100mLとする。
塩化第二銅 CuCl2・2H2O〔特級〕
塩化チオニル SOCl2〔特級〕
塩化ナトリウム NaCl〔特級〕
塩化ナトリウム(標準試薬) NaCl〔容量分析用標準試薬〕
塩化ナトリウム試液 塩化ナトリウム10g(9.5~10.4g)に水を加えて溶かし、100mLとする。
塩化ナトリウム・水酸化ナトリウム試液 塩化ナトリウム100g(99.5~100.4g)に2.5mol/L水酸化ナトリウム試液40mL及び水を加えて溶かし、1,000mLとする。
塩化バリウム BaCl2・2H2O〔特級〕
塩化バリウム試液 塩化バリウム12g(11.5~12.4g)に水を加えて溶かし、100mLとする(0.5mol/L)。
塩化ベンザルコニウム 塩化ベンザルコニウム〔日局〕
塩化マグネシウム MgCl2・6H2O〔特級〕
塩化マグネシウム試液 酸化マグネシウム3.75g(3.745~3.754g)に8mol/L塩酸試液を少量ずつ加えて溶かし、100mLとする。
塩化メチルロザニリン C25H30ClN3〔日局〕
塩化メチルロザニリン・エタノール試液、0.25mol/L 塩化メチルロザニリン10g(9.5~10.4g)をエタノール100mLに溶かす。
塩化メチルロザニリン・エタノール試液、0.037mol/L 塩化メチルロザニリン0.3g(0.25~0.34g)をエタノール20mLに溶かす。
塩化メチルロザニリン試液 塩化メチルロザニリン0.1g(0.05~0.14g)に氷酢酸10mLを加えて溶かす。
塩化リチウム LiCl・H2O〔アミノ酸分析用〕
塩基性フクシン 〔特級〕
塩基性フクシン試液 塩基性フクシン10g(9.5~10.4g)をエタノール100mLに溶かし、37℃で一夜放置する。
塩酸 HCl〔特級〕 35.0%以上。
塩酸、希 塩酸23.6mLに水を加えて100mLとする(10%)。
塩酸グアニン C5H5N5O・HCl 白色の結晶又は結晶性の粉末である。希塩酸に溶け、水又はエタノールにほとんど溶けない。
溶状 本品0.2g(0.15~0.24g)に塩酸(2→3)20mLを加え、加温溶解するとき、溶液は、ほとんど澄明である。
水分 5.0%以下(1g)
含量 94.0%以上
塩酸試液、8mol/L 塩酸720mLに水を加えて1,000mLとする。
塩酸試液、6mol/L 塩酸540mLに水を加えて1,000mLとする。
塩酸試液、3mol/L 塩酸270mLに水を加えて1,000mLとする。
塩酸試液、2.5mol/L 塩酸225mLに水を加えて1,000mLとする。
塩酸試液、2mol/L 塩酸180mLに水を加えて1,000mLとする。
塩酸試液、1mol/L 塩酸90mLに水を加えて1,000mLとする。
塩酸試液、0.5mol/L 塩酸45mLに水を加えて1,000mLとする。
塩酸試液、0.2mol/L 1mol/L塩酸試液200mLに水を加えて1,000mLとする。
塩酸試液、0.1mol/L 1mol/L塩酸試液100mLに水を加えて1,000mLとする。
塩酸試液、0.001mol/L 0.1mol/L塩酸試液10mLに水を加えて1,000mLとする。
塩酸システイン HSCH2CH(NH2)COOH・HCl〔特級〕
塩酸ジメチルアミン (CH3)2NH・HCl 白色の結晶で潮解性があり、水には極めてよく溶ける。
融点 170~172℃
塩酸チアミン C12H17ClN4OS・HCl〔日局〕
塩酸L-ヒスチジン、定量用 乾燥したものを定量するとき、塩酸L-ヒスチジン(C6H9N302・HCl・H2O)98.5%以上を含むもの。
塩酸ヒドロキシルアミン NH2OH・HCl〔特級〕
塩酸ピリドキシン C8H11NO3・HCl〔日局〕
0.1mol/L塩酸・メタノール試液 塩酸9.0mLにメタノールを加えて1,000mLとする。
0.01mol/L塩酸・メタノール試液 1mol/L塩酸試液10mLにメタノールを加えて1,000mLとする。
塩酸、誘導結合プラズマ分析用 HCl〔微量金属分析用、35.0~37.0%〕
塩酸リジン C6H14N2O2・HCl〔日局〕
塩素 Cl2 窒息性で、臭いがある黄緑色のガスで、空気より重く、水に溶ける。サラシ粉に塩酸を作用させて調製する。塩素ボンベに入れたものを用いることができる。
黄色酸化第二水銀 酸化第二水銀、黄色の項に定める。
王水 塩酸3容量に硝酸1容量を加える。用時調製する。
2-オキシ-1-(2′-オキシ-4′-スルホ-1′-ナフチルアゾ)-3-ナフトエ酸 C21H14N2O7S〔2-ヒドロキシ-1-(2′-ヒドロキシ-4′-スルホ-1′-ナフチルアゾ)-3-ナフトエ酸、特級〕
8-オキシキノリン C9H6NOH〔8-キノリノール、特級〕
8-オキシキノリン試液 8-オキシキノリン20mg(19.5~20.4mg)を水酸化ナトリウム溶液(13→100)100mLに溶かす。
1-オクタンスルホン酸ナトリウム C8H17NaO3S 本品は、白色の粉末である。
溶状 本品1.1g(1.05~1.14g)を量り、水50mLを加えて溶かした液は澄明である。
含量 98.0%以上
定量法 105℃で2時間乾燥した本品約0.4gを0.0001gの桁まで量り、その数値を記録し、水25 mLを加え、0.1mol/L水酸化ナトリウム溶液で滴定する(指示薬 フェノールフタレイン溶液2~3滴)。終点は、液の色が微赤色を15秒間保つときとする。
0.1mol/L水酸化ナトリウム溶液1mL=21.672mg CH3(CH2)7SO3Na
オクタン酸 CH3(CH2)6COOH 〔アミノ酸分析用〕
オクチルフェノールエトキシレート C14H22O(C2H4O)n
オクチルフェノールエトキシレート試液 オクチルフェノールエトキシレート25g(24.5~25.4g)に水を加えて溶かし250mLとする。
オリブ油 〔日局〕
過塩素酸 HClO4〔特級、比重約1.67〕 HClO470~72%を含む。
過塩素酸第二鉄 Fe(ClO4)3 灰白色~淡褐色の結晶で潮解性がある。水に極めて溶けやすい。
溶状 本品の水溶液(1→20)は、澄明である。
塩化物 本品0.20g(0.195~0.204g)を量り、塩化物の試験を行うとき、その量は、0.001mol/L塩酸0.40mLに対応する量以下でなければならない(0.071%以下)。
過塩素酸第二鉄試液 過塩素酸第二鉄3g(2.5~3.4g)に水を加えて溶かし、500mLとし、ろ過する。
カザミノ酸 ビタミン類不含のカゼインを塩酸酸性で加水分解し、適当な処理により粉末としたものである。白色~淡黄色の粉末で、特異な臭いがある。
過酸化水素試液 強過酸化水素水1容量に水9容量を加える(3%)。用時調製する。
過酸化水素水、強 H2O2〔過酸化水素水(30%)、特級〕 H2O230w/v%以上を含む。冷暗所に保存する。
カゼイン、乳製 白色~ほとんど白色の粉末である。
溶状 本品0.6g(0.55~0.64g)に0.05mol/L乳酸試液100mLを加えて溶かすとき、溶液は、微濁以下である。
吸光度 本品0.6gを0.001gの桁まで量り、その数値を記録し、0.05mol/L乳酸試液に溶かし、100mLとし、この溶液につき、波長400nmで吸光度を測定するとき、E1cm0.6%≦0.4である。
カゼイン試液 乳製カゼイン0.1g(0.05~0.14g)に水30mLを加え、よく分散させた後、水酸化ナトリウム溶液(1→10)1mLを加えて溶かし、更に水を加えて50mLとする。用時調製する。
カゼイン製ペプトン ペプトン、カゼイン製の項に定める。
活性炭 〔日局〕
カテコール C6H4(OH)2〔1級〕
カフェイン C8H10N4O2・H2O〔日局〕
過マンガン酸カリウム KMnO4〔特級〕
過マンガン酸カリウム試液 過マンガン酸カリウム3.3g(3.25~3.34g)に水を加えて溶かし、1,000mLとする(0.02mol/L)。
過ヨウ素酸 HIO4〔メタ過ヨウ素酸、特級〕
過ヨウ素酸試液 過ヨウ素酸2.5g(2.45~2.54g)に水を加えて溶かし、100mLとした後、氷酢酸400mLを加える。遮光して保存する。
過ヨウ素酸カリウム KIO4〔特級〕
過ヨウ素酸ナトリウム試液 メタ過ヨウ素酸ナトリウム25g(24.5~25.4g)に水に加えて溶かし、100mLとする。
ガラス繊維 〔ガラスウール、特級〕
過硫酸アンモニウム (NH4)2S2O8〔特級〕
カールフィッシャー試液 一般試験法の水分定量法に定めるところによるものとする。
カールフィッシャー用エチレングリコール エチレングリコール、カールフィッシャー用の項に定める。
カールフィッシャー用ピリジン 一般試験法の水分定量法に定めるところによるものとする。
カールフィッシャー用メタノール 一般試験法の水分定量法に定めるところによるものとする。
カルボキシメチルセルロースナトリウム 〔日局〕 置換度が0.62~0.68のものに限る。
緩衝液用0.2mol/Lリン酸二水素カリウム試液 0.2mol/Lリン酸二水素カリウム試液、緩衝液用の項に定める。
肝臓エキス 茶褐色の粉末又は粒子である。
乾燥減量 6%以下(1g,85℃,1時間)
溶解性 本品の溶液(1→1,000)をpH7.0に調整し、121℃で15分間高圧蒸気滅菌するとき、不溶物を認めない。
カンテン 〔日局〕
希エタノール、エタノール、希の項に定める。
希塩化第二鉄試液 塩化第二鉄試液、希の項に定める。
希塩酸 塩酸、希の項に定める。
希酢酸 酢酸、希の項に定める。
ギ酸 HCOOH〔特級〕
ギ酸ナトリウム、定量用 HCOONa〔特級〕
希2,4-ジニトロクロルベンゼン試液 2,4-ジニトロクロルベンゼン試液、希の項に定める。
希硝酸 硝酸、希の項に定める。
キシラン 植物繊維をアルカリで抽出して得られる複雑な多糖類で、加水分解によりキシロースを生ずる。5℃以下で保存する。
キシレンシアノールFF C25H27N2NaO7S2〔1級〕
キシロース C5H10O5 白色~微褐色の結晶性粉末又は粉末である。
溶状 本品1g(0.5~1.4g)に水20mLを加えて溶かすとき、溶液は、澄明又はほとんど澄明である。
比旋光度 本品20gを0.1gの桁まで量り、その数値を記録し、水を加えて溶かし、100mLの全量フラスコに入れ、水を標線まで加えて100mLとし、この溶液につき、層長100mmで旋光度を測定するとき、〔α〕D20=+18.0~+20.0°である。
乾燥減量 0.3%以下(1g,105℃,2時間)
強熱残分 0.1%以下(1g)
希水酸化ナトリウム試液 水酸化ナトリウム試液、希の項に定める。
希水酸化ナトリウム・エタノール試液 水酸化ナトリウム・エタノール試液、希の項に定める。
キナルジンレッド(C21H23N2I) 本品は、結晶性粉末でエタノールに溶けやすい。本品のメタノール溶液(0.005→1,000)は、526nm付近に極大吸収部がある。また、当該極大吸収部で吸光度を測定するとき、0.5以上である。
キナルジンレッド試液 キナルジンレッド0.1g(0.05~0.14g)を量り、酢酸100mLを加えて溶かす。用時調製する。
強アンモニア水 アンモニア水、強の項に定める。
強過酸化水素水 過酸化水素水、強の項に定める。
強酸性陽イオン交換樹脂 スルホン酸基が導入してある架橋度8%のスチレン-ジビニルベンゼン共重合体を、高速液体クロマトグラフ用に製造したものとする。
希硫酸 硫酸、希の項に定める。
金属ナトリウム ナトリウム、金属の項に定める。
クエン酸 C6H8O7・H2O〔特級〕
クエン酸、無水 C6H8O7〔日局〕
クエン酸アンモニウム C6H14N2O7〔クエン酸二アンモニウム、特級〕
0.2mol/Lクエン酸塩緩衝液、pH5.2 クエン酸ナトリウム59.6g(59.55~59.64g)、クエン酸21.8g(21.75~21.84g)に水約750mLを加えて溶かし、必要ならば、クエン酸溶液(1→50)又はクエン酸ナトリウム溶液(1→100)を用いてpH5.2に調整した後、水を加えて1,000mLとする。
クエン酸塩緩衝液、酵素力試験用 0.1mol/L塩酸試液に0.1mol/Lクエン酸二ナトリウム試液を加えて所定のpHに調整する。
クエン酸緩衝液 クエン酸21.0g(20.95~21.04g)及び水酸化ナトリウム8.4g(8.35~8.44g)に水約700mLを加えて溶かし、塩酸でpH1.3に調整した後、水を加えて1,000mLとする。
クエン酸ナトリウム Na3C6H5O7・2H2O〔特級〕
クエン酸ナトリウム緩衝液 クエン酸ナトリウム980g(979.5~980.4g)に水3,500mL、塩酸700mL及びオクタン酸5mLを加え、1mol/L塩酸でpH2.2に調整した後、水を加えて5,000mLとする。この溶液500mLに20v/v%チオジエチレングリコール溶液500mL及び水3,500mLを加え、1mol/L塩酸でpH2.2に調整した後、水を加えて5,000mLとする。
クエン酸二ナトリウム C6H6Na2O7・1(1/2)H2O〔特級〕
クエン酸二ナトリウム試液、0.1mol/L クエン酸二ナトリウム26.3g(26.25~26.34g)に水を加えて1,000mLとする。
クエン酸リチウム Li3C6H5O7・4H2O 〔アミノ酸分析用〕
クエン酸リチウム緩衝液 クエン酸リチウム6.9g(6.85~6.94g)、塩化リチウム1.3g(1.25~1.34g)、クエン酸8.8g(8.75~8.84g)、塩酸4.0mL、エタノール40.0mL、チオジエチレングリコール2.5mL及びオクタン酸0.1mLに水を加えて溶かし、1,000mLとし、塩酸及び5mol/L水酸化リチウムでpHを2.98に調整する。
クリスタルバイオレット 〔特級〕
クリスタルバイオレット・氷酢酸試液 クリスタルバイオレット50mg(49.5~50.4mg)を氷酢酸100mLに溶かす。
グリセリン C3H8O3〔濃グリセリン、日局〕
β-グルカン 大麦、えん麦、酵母等の細胞壁に存在する多糖類で、加水分解によりD-グルコースを生ずる。ただし、大麦由来の分子量約200,000のものを用いる。
m-クレゾール CH3C6H4(OH)〔1級〕
クロム酸カリウム K2CrO4〔特級〕
クロム酸カリウム試液 クロム酸カリウム10g(9.5~10.4g)に水を加えて溶かし、100mLとする。
クロマトグラフ用ケイソウ土 セイソウ土、クロマトグラフ用の項に定める。
クロマトグラフ用DEAE-セファデックスA-25 DEAE-セファデックスA-25、クロマトグラフ用の項に定める。
クロモトロプ酸 (HO)2C10H4(SO3Na)2〔クロモトロプ酸(二ナトリウム塩)、特級〕 遮光して保存する。
クロモトロプ酸試液 水30mLに硫酸68mLを注意して加え、放冷した後、水を加えて100mLとした溶液にクロモトロプ酸0.05g(0.045~0.054g)を加えて溶かす。遮光して保存する。
クロラミン C7H7ClNNaO2S・3H2O〔クロラミンT、特級〕
クロラミン試液 クロラミン1g(0.5~1.4g)に水を加えて溶かし、100mLとする。用時調製する。
クロラムフェニコール C11H12Cl2N2O5〔日局〕
p-クロロフェノール ClC6H4OH 無色~僅かに赤色の結晶又は結晶の塊で特異な臭いがある。エタノール、クロロホルム、エーテル又はグリセリンに極めて溶けやすく、水にやや溶けにくい。
融点 約43℃
含量 99.0%以上
定量法 本品約0.2gを0.001gの桁まで量り、その数値を記録し、水を加えて溶かし、100mLの全量フラスコに入れ、更に水を標線まで加えて100mLとする。この溶液25mLを全量ピペットを用いて量り、ヨウ素瓶に入れ、0.05mol/L臭素溶液20mLを全量ピペットを用いて加え、さらに、塩酸5mLを加え、30秒以内に密栓し、30分間しばしば振り混ぜ、さらに、15分間放置する。次に、ヨウ化カリウム溶液(1→5)5mLを加え、30秒以内に密栓し、よく振り混ぜた後、0.1mol/Lチオ硫酸ナトリウム溶液で滴定する(指示薬 デンプン試液1mL)。同様の方法で空試験を行う。
0.05mol/L臭素溶液1mL=3.214mgC6H5ClO
クロロホルム CHCl3〔特級〕
クロロホルム、エタノール不含 クロロホルム20mLを水20mLと3分間穏やかによく振り混ぜた後、クロロホルム層を分取し、さらに、水20mLずつで2回洗い、乾燥ろ紙でろ過し、無水硫酸ナトリウム5g(4.5~5.4g)を加え、5分間よく振り混ぜ、2時間放置した後、乾燥ろ紙でろ過する。用時調製する。
ケイソウ土、クロマトグラフ用 白色~灰白色の上質のものを用いる。
酵母エキス 適当な条件下で酵母(Saccharomyces)の産出物のペプトン様の総水溶性物質を澄明液とし、蒸発乾燥し、粉末としたもので、本品1gは、原料酵母7.5g以上から得たものである。帯赤黄色~褐色の粉末で腐敗臭のない特異な臭いがある。水に溶けて黄色~褐色の弱酸性の溶液となる。本品には、特別に含水炭素を加えない。
凝固性たん白質 本品の水溶液(1→20)を沸騰するまで加熱するとき、沈殿を生じない。
塩化物(NaClとして) 5.0%以下
乾燥減量 5.0%以下(0.5g,105゚,恒量)
強熱残分 15.0%以下(0.5g)
窒素含量 7.2から9.5%(105゚,恒量,乾燥後,窒素定量法による。)
五酸化リン P2O5〔特級〕
コバルチ亜硝酸ナトリウム Na3Co(NO2)6〔特級〕
コバルト亜硝酸ナトリウム試液 コバルト亜硝酸ナトリウム10g(9.5~10.4g)に水を加えて溶かし、50mLとする。必要ならば、ろ過する。用時調製する。
酢酸 CH3COOH〔日局〕(5mol/L)
酢酸亜鉛 Zn(CH3COO)2・2H2O〔特級〕
酢酸イソアミル CH3COOCH2CH2CH(CH3)2〔特級〕
酢酸、希 氷酢酸6g(5.5~6.4g)に水を加えて100mLとする(1mol/L)。
酢酸、氷 CH3COOH〔酢酸(氷酢酸)(99~100%)、特級〕
酢酸、氷、非水滴定用 氷酢酸1Lに三酸化クロム5g(4.5~5.4g)を加え、一夜放置した後、ろ過して蒸留し、115℃以上の留分に無水酢酸20g(19.5~20.4g)を加え、再蒸留し、117~118℃で定沸点になった留分をとる。
酢酸アンモニウム CH3COONH4〔特級〕
酢酸アンモニウム試液 酢酸アンモニウム10g(9.5~10.4g)に水を加えて溶かし、100mLとする。
酢酸・エタノール試液 氷酢酸1mLに水9mLを加え、さらに、エタノール10mLを加え、混和する。
酢酸エチル CH3COOC2H5〔特級〕
酢酸塩酸緩衝液、0.1mol/L、酵素力試験用 0.1mol/L酢酸ナトリウム試液に0.1mol/L塩酸試液を加えて所定のpHに調整する。
酢酸・酢酸アンモニウム緩衝液、pH4.8 酢酸アンモニウム77g(76.5~77.4g)に水約200mLを加えて溶かし、これに氷酢酸57mLを加え、水を加えて1,000mLとする。
酢酸・酢酸ナトリウム緩衝液、pH5.0 酢酸ナトリウム試液140mLに希酢酸60mL及び水を加えて1,000mLとする。
酢酸・酢酸ナトリウム緩衝液、1mol/L、酵素力試験用 酢酸ナトリウム試液に希酢酸を加えて所定のpHに調整する。
酢酸・酢酸ナトリウム緩衝液、0.2mol/L、酵素力試験用 0.2mol/L酢酸ナトリウム試液に0.2mol/L酢酸試液を加えて所定のpHに調整する。
酢酸・酢酸ナトリウム緩衝液、0.1mol/L、酵素力試験用 0.2mol/L酢酸ナトリウム試液に0.2mol/L酢酸試液を加えて所定のpHに調整し、水を加えて正確に2倍容量とする。
酢酸・酢酸ナトリウム緩衝液、0.01mol/L、酵素力試験用 0.2mol/L酢酸ナトリウム試液に0.2mol/L酢酸試液を加えて所定のpHに調整し、水を加えて正確に20倍容量とする。
酢酸・酢酸ナトリウム緩衝液、0.005mol/L、酵素力試験用 0.2mol/L酢酸ナトリウム試液に0.2mol/L酢酸試液を加えて所定のpHに調整し、水を加えて正確に40倍容量とする。
酢酸・酢酸リチウム緩衝液 酢酸リチウム407g(406.5~407.4g)及び酢酸200mLに水を加えて溶かし、2,000mLとする。
酢酸試液、6mol/L 氷酢酸36g(35.5~36.4g)に水を加えて溶かし、100mLとする。
酢酸試液、1mol/L 氷酢酸12g(11.5~12.4g)に水を加えて溶かし、200mLとする。
酢酸試液、0.2mol/L 氷酢酸12g(11.5~12.4g)に水を加えて溶かし、1,000mLとする。
酢酸スチグマステロール 一般試験法のビタミンD定量法に定めるところによるものとする。
酢酸第二水銀 Hg(CH3COO)2〔特級〕
酢酸第二水銀試液、非水滴定用 酢酸第二水銀5g(4.5~5.4g)に非水滴定用氷酢酸を加えて溶かし、100mLとする。
酢酸dl -α-トコフェロール、定量用 酢酸dl -α-トコフェロール製造用原体。ただし、定量するとき、酢酸dl -α-トコフェロール98.0%以上のものに限る。
酢酸ナトリウム CH3COONa・3H2O〔特級〕
酢酸ナトリウム、無水 CH3COONa〔酢酸ナトリウム(無水)、特級〕
0.25mol/L酢酸ナトリウム塩酸混液(pH5.5) 酢酸ナトリウム34.02g(34.015~34.024g)に水を加えて溶かし、塩酸でpHを5.5に調整し、水を加えて1,000mLとする。
0.25mol/L酢酸ナトリウム緩衝液(pH5.5) 酢酸ナトリウム30.02g(30.015~30.024g)に水約900mLを加えて溶かし、氷酢酸1.68mLをマイクロピペットを用いて加え、塩化カルシウム0.147g(0.1465~0.1474g)を加え、更にポリソルベート20試液1mLを全量ピペットを用いて加え、1mol/L酢酸試液又は1mol/L水酸化ナトリウム試液を用いてpHを5.5に調整し、水を加えて1,000mLとする。
酢酸ナトリウム試液 酢酸ナトリウム13.6g(13.55~13.64g)に水を加えて溶かし、100mLとする(1mol/L)。
酢酸ナトリウム試液、0.2mol/L 酢酸ナトリウム27.2g(27.15~27.24g)に水を加えて溶かし、1,000mLとする。
酢酸ナトリウム試液、0.1mol/L 酢酸ナトリウム試液100mLに水を加えて1,000mLとする。
酢酸鉛 Pb(CH3COO)2・3H2O〔特級〕
酢酸鉛試液 酢酸鉛9.5g(9.45~9.54g)に、新たに煮沸し冷却した水を加えて溶かし、100mLとする。密栓して保存する(0.25mol/L)。
酢酸4-ニトロフェニル CH3COOC6H4NO2 白色~淡黄色の結晶性の粉末である。
融点 77~80℃
モル吸光係数 本品約200mgを1mgの桁まで量り、その数値を記録し、メタノールを加えて溶かし、100mLの全量フラスコに入れ、更にメタノールを標線まで加えて100mLとする。この溶液10mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、メタノールを標線まで加えて100mLとし、さらに、この溶液5mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、メタノールを標線まで加えて100mLとする。この溶液につき、メタノールを対照液として波長268nmにおける吸光度を測定するとき、本品のモル吸光係数は、9,200~9,600である。
酢酸n-ブチル CH3COOCH2CH2CH2CH3〔特級〕
酢酸、無水 無水酢酸の項に定める。
酢酸リチウム CH3COOLi・2H2O 〔アミノ酸分析用〕
サフラニン 本品は、類黒色の粉末又は小塊で、水及びエタノールにやや溶けにくい。
溶状 本品0.1g(0.05~0.14g)を水100mLに溶かし、水浴上で加熱溶解するとき、溶液の色は、ほとんど澄明である。
吸光度 本品0.1gを0.001gの桁まで量り、その数値を記録し、水を加えて溶かし、100mLの全量フラスコに入れ、更に水を標線まで加えて100mLとし、試料原液とする。試料原液を水で正確に200倍希釈して試料溶液とする。波長517nmにおける吸光度を測定するとき、0.45~0.65の値を得る。
乾燥減量 5%以下(1g,105℃,4時間)
強熱残分 1.0%以下
三塩化アンチモン SbCl3〔特級〕
三塩化アンチモン試液 クロロホルムを等容量の水で2~3回洗い、新たに強熱して冷却した炭酸カリウムを加え、密栓し、遮光して一夜放置し、クロロホルム層を分取し、できる限り遮光して蒸留する。このクロロホルムで三塩化アンチモンの表面を洗い、洗液が澄明となったとき、クロロホルムを加えて飽和溶液とし、遮光した共栓瓶に入れる。用時調製する。
残留農薬試験用ベンゼン ベンゼン、残留農薬試験用
酸化第二水銀、黄色 HgO〔酸化第二水銀(黄色)、特級〕 遮光して保存する。
酸化バリウム BaO〔乾燥用〕
酸化マグネシウム MgO〔特級〕
三酸化クロム CrO3〔三酸化クロム(無水クロム酸)、特級〕
三酸化ヒ素(標準試薬)〔容量分析用標準試薬〕
酸性塩化カリウム試液 塩化カリウム250g(249.5~250.4g)に水を加えて溶かし、1,000mLとした溶液に塩酸8.5mLを加える。
酸性塩化第一スズ試液 塩化第一スズ試液、酸性の項に定める。
次亜塩素酸ナトリウム試液 次亜塩素酸ナトリウム(NaClO:74.44)が5%含量となるように、1mol/L水酸化ナトリウム試液に氷冷しながら塩素を通じて調製する。用時調製する。
次亜塩素酸ナトリウム試液、アンモニウム試験用 水酸化ナトリウム又は炭酸ナトリウムの水溶液に塩素を吸収させた無色~淡緑黄色で、澄明の溶液である。
含量 次亜塩素酸ナトリウム(NaClO:74.44)として4.2g/dL以上
定量法 本品10mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、水を標線まで加えて100mLとする。この溶液10mLを全量ピペットを用いて共栓フラスコに入れ、水90mLを加えた後、ヨウ化カリウム2g(1.5~2.4g)及び酢酸(1→2)6mLを加え、密栓し、よく振り混ぜ、暗所に5分間放置する。遊離したヨウ素を、0.1mol/Lチオ硫酸ナトリウム溶液で滴定する(指示薬 デンプン試液3mL)。同様の方法で空試験を行い補正する。
0.1mol/Lチオ硫酸ナトリウム溶液1mL=3.722mgNaClO
ジアゾベンゼンスルホン酸試液 105℃で3時間乾燥したスルファニル酸0.9g(0.85~0.94g)を量り、希塩酸10mLを加え、加熱して溶かし、水を加えて100mLとする。この溶液3.0mLを量り、亜硝酸ナトリウム試液25mLを加え、氷冷しながら5分間放置した後、亜硝酸ナトリウム試液5mL及び水を加えて100mLとし、氷水中で15分間放置する。用時調製する。
5-シアノアンミン鉄ナトリウム ナトリウムペンタシアノアンミンフェロエートの項に定める。
次亜リン酸 H3PO2〔1級〕 H3PO230~32%を含む。
シアン化カリウム KCN〔特級〕
シアン化カリウム試液 シアン化カリウム1g(0.5~1.4g)に水を加えて溶かし、10mLとする。用時調製する。
シアン酢酸エチル NCCH2COOC2H5〔1級〕
ジエチルジチオカルバミン酸銀 C5H10AgNS2〔日局〕
四塩化炭素 CCl4〔特級〕
ジオキサン C4H8O2〔特級〕
2,7-ジオキシナフタリン C10H6(OH)2(2,7-ジヒドロキシナフタリン) 白色針状の結晶又は結晶性の粉末で、エタノール又はエーテルに溶けやすく、水に溶けにくい。
融点 190℃
2,7-ジオキシナフタリン試液 2,7-ジオキシナフタリン0.025g(0.0245~0.0254g)をメタノール1,000mLに溶かし、この溶液90mLにフェリシアン化カリウム溶液(1→500)5mL及びシアン化カリウム溶液(1→100)5mLを加え、混和した後、30分間放置し、水酸化ナトリウム溶液(2.25→200)15mLにメタノールを加えて200mLとした溶液100mLを加える。用時ろ過し、75分以内に使用する。
ジギトニン C55H90O29 白色の結晶性の粉末で、水に溶けにくい。温エタノール又は氷酢酸に溶ける。クロロホルム又はエーテルに溶けない。
溶状 本品0.5g(0.45~0.54g)に温エタノール20mLを加えて溶かすとき、溶液は、無色で、澄明である。
融点 約230℃(分解)
比旋光度〔α〕D20=-47~-49°(1g,75%酢酸,10mL,100mm)
乾燥減量 6%以下(105℃)
強熱残分 0.3%以下
β-シクロデキストリン (C6H10O5)7 白色の結晶性の粉末である。
乾燥減量 12.0%以下(1g,105℃,3時間)
強熱残分 8.2%以下(1g)
β-シクロデキストリン緩衝液 β-シクロデキストリン5.5g(5.45~5.54g)に一般試験法の抗生物質の力価試験法に規定する3号緩衝液を加えて溶かし、1,000mLとする。
シクロヘキサノン C6H10O〔特級〕
シクロヘキサン C6H12〔特級〕
2,6-ジクロルフェノールインドフェノールナトリウム C12H6Cl2NNaO2〔特級〕
2,6-ジクロルフェノールインドフェノールナトリウム試液 2,6-ジクロルフェノールインドフェノールナトリウム0.1g(0.05~0.14g)に水100mLを加え、加温した後、ろ過する。3日以内に使用する。
2,6-ジクロルフェノールインドフェノールナトリウム試液、滴定用 炭酸水素ナトリウム0.042g(0.0415~0.0424g)に水50mLを加えて溶かし、さらに、2,6-ジクロルフェノールインドフェノールナトリウム0.05g(0.045~0.054g)を溶かし、水を加えて200mLとし、ろ過する。用時調製する。
ジクロロメタン CH2Cl2〔特級〕
次酢酸鉛試液 酢酸鉛3g(2.5~3.4g)及び一酸化鉛1g(0.5~1.4g)に水0.5mLを加え、すり混ぜ、得られた類黄色の混和物をビーカーに入れ、時計皿で覆い、水浴上で加熱し、均等の白色又は帯赤白色になったとき、さらに、熱湯9.5mLを少量ずつ加え、再び時計皿で覆い、放置した後、上澄液を傾斜してとり、水を加えてその比重を1.23~1.24(15℃)に調整する。密栓して保存する。
四シュウ酸カリウム、pH測定用 KH3(C2O4)2・2H2O〔pH測定用〕
次硝酸ビスマス 〔日局〕
L-シスチン HOOC(NH2)CHCH2SSCH2CH(NH2)COOH〔特級〕
L-システイン塩酸塩一水和物 HSCH2CH(NH2)COOH・HCl・H2O〔特級〕
ジチゾン C6H5NHNHCSN:NC6H5〔ジチゾン(ジフェニルチオカルバゾン)、特級〕
ジチゾン試液 ジチゾン25mg(24.5~25.4mg)を量り、エタノール100mLを加えて溶かす。用時調製する。
3,5-ジニトロ塩化ベンゾイル (NO2)2C6H3COCl〔特級〕
2,4-ジニトロクロルベンゼン C6H3(NO2)2Cl〔特級〕
2,4-ジニトロクロルベンゼン試液 2,4-ジニトロクロルベンゼン0.01g(0.005~0.014g)を量り、残留農薬試験用ベンゼンを加えて溶かし、100mLの全量フラスコに入れ、更に残留農薬試験用ベンゼンを標線まで加えて100mLとする。この溶液1mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、残留農薬試験用ベンゼンを標線まで加えて100mLとする。
2,4-ジニトロクロルベンゼン試液、希 2,4-ジニトロクロルベンゼン試液10mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、残留農薬試験用ベンゼンを標線まで加えて100mLとする。
3,5-ジニトロサリチル酸 (O2N)2C6H2(OH)COOH 僅かに薄い黄色~薄い黄褐色の結晶性粉末又は粉末で、エタノール及びアセトンに溶けやすく、水に溶けにくい。
溶状 本品1g(0.5~1.4g)にエタノール20mLを加えて溶かすとき、溶液は、ほとんど透明である。
融点 171~175℃
確認試験 本品1mg(0.5~1.4mg)を量り、赤外吸収スペクトル測定法の臭化カリウム錠剤法により赤外吸収スペクトルを測定するとき、波数3,100cm-1、1,680cm-1、1,600cm-1、1,540cm-1、1,340cm-1、1,220cm-1、1,160cm-1、1,090cm-1、900cm-1、810cm-1、740cm-1及び710cm-1付近に主な吸収を認める。
含量 98.0%以上
定量法 本品0.4gを0.001gの桁まで量り、その数値を記録し、エタノール10mL及び水20mLを加え、0.1mol/L水酸化ナトリウム溶液で滴定する(電位差滴定法)。
0.1mol/L水酸化ナトリウム溶液1mL=0.02281g(O2N)2C6H2(OH)COOH
3,5-ジニトロサリチル酸試液 3,5-ジニトロサリチル酸20.0g(19.95~20.04g)に水800mLを加え、懸濁し、かき混ぜながら酵素力試験用水酸化ナトリウム試液300mLを少量ずつ加える。これを48℃以下の水浴中で加温し、溶液が透明になるまでかき混ぜる。これに、酒石酸カリウムナトリウム600g(599.5~600.4g)を少量ずつ加え、溶液が透明になるまでかき混ぜる。必要ならば、これを48℃以下の水浴中で加温し、かき混ぜる。さらに、水を加えて2,000mLとし、ガラスろ過器(G3)でろ過し、遮光した容器に保存する。180日以内に使用する。
α,α′-ジピリジル C10H8N2〔特級〕
α,α′-ジピリジル試液 α,α′-ジピリジル0.25g(0.245~0.254g)にエタノールを加えて溶かし、100mLとする。用時調製する。
ジフェニルアミン (C6H5)2NH〔特級〕
ジフェニルアミン試液 ジフェニルアミン1g(0.5~1.4g)に硫酸100mLを加えて溶かす。無色の溶液を用いる。
ジフェニルカルバゾン C6H5NHNHCON:NC6H5〔特級〕
ジフェニルカルバゾン試液 ジフェニルカルバゾン1g(0.5~1.4g)にエタノールを加えて溶かし、1,000mLとする。
ジフェニルチオカルバゾン C6H5NHNHCSN:NC6H5〔ジチゾン、特級〕
2,6-ジブロムキノンクロルイミド O:C6H2Br2:NCl〔特級〕
ジベンゾイルチアミン定量用臭化シアン試液 臭化シアン試液、ジベンゾイルチアミン定量用の項に定める。
p-ジメチルアミノシンナムアルデヒド C11H13NO 橙色の結晶又は結晶性の粉末で、特異な臭いがある。希塩酸に溶けやすく、エタノール又はエーテルに溶けにくく、水にほとんど溶けない。
溶状 本品0.2g(0.15~0.24g)にエタノール20mLを加えて溶かすとき、溶液は、澄明である。
融点 140~142℃
乾燥減量 0.5%以下(1g,105℃,2時間)
強熱残分 0.10%以下(1g)
窒素含量 7.8~8.1%(105℃,2時間,乾燥した後,窒素定量法によるものとする。)
p-ジメチルアミノシンナムアルデヒド試液 p-ジメチルアミノシンナムアルデヒドのエタノール溶液(1→2,000)10mLに、用時、氷酢酸1mLを加える。
p-ジメチルアミノベンズアルデヒド (CH3)2N・C6H4CHO〔特級〕
p-ジメチルアミノベンズアルデヒド・エタノール硫酸試液 p-ジメチルアミノベンズアルデヒド1.5g(1.45~1.54g)に約50mLの無水エタノールを加えて溶かした後、硫酸0.5mLを加えて無水エタノールで100mLとする。用時調製する。
p-ジメチルアミノベンズアルデヒド・塩化第二鉄試液 p-ジメチルアミノベンズアルデヒド125mg(124.5~125.4mg)を硫酸65mL及び水35mLの冷混液に溶かし、塩化第二鉄試液0.05mLを加える。調製した後7日以内に用いる。
ジメチルスルホキシド CH3SOCH3〔特級〕
ジメチルホルムアミド HCON(CH3)2〔N,N-ジメチルホルムアミド、特級〕
弱酸性陽イオン交換樹脂 メタアクリル酸ジビニルベンゼン共重合体を液体クロマトグラフ用に製造した上質のものとする。
弱酸性陽イオン交換樹脂カラム 弱酸性陽イオン交換樹脂を0.5mol/L硫酸試液中に分散させ、3時間放置する。洗液が中性になるまで水で洗った後、かき混ぜながらpHを7~8に保つように水酸化リチウムを加える。pHが安定した後、一夜放置し、水で5回以上洗い、リン酸溶液(1→25)でpHを7.0に調整し、保存する。用時、内径1cmのカラムに高さ10cmとなるよう充填し、試料溶液を負荷する直前に水25mL以上で洗う。
弱石炭酸フクシン液 塩基性フクシン試液10mLをフェノール溶液(5→100)100mLに加え、さらに、水で10~20倍に希釈する。用時調製する。
臭化カリウム KBr〔特級〕
臭化カリウム、赤外用 臭化カリウム単結晶又は臭化カリウム〔特級〕を砕き、200号(74μm)ふるいを通過したものを集め、120℃で10時間又は500℃で5時間乾燥した粉末である。これを用いて錠剤を作るとき、異常な吸収を認めてはならない。
臭化シアン試液 氷冷した水100mLに臭素1mLを加え、激しく振り混ぜた後、氷冷したシアン化カリウム試液を臭素の色が脱色するまで滴加する。この試液は、ドラフトの中で用時調製する。
この試液の蒸気は、極めて有毒であるから、取扱いに際し、吸入しないよう注意する。
臭化シアン試液、ジベンゾイルチアミン定量用 氷冷した水100mLに臭素2mLを加え、激しく振り混ぜた後、氷冷したチオシアン酸カリウム試液を臭素の色が脱色するまで滴加する。この試液は、ドラフトの中で調製し、冷所に保存する。調製した後1か月以内に用いる。
この試液の蒸気は極めて有毒であるから取扱いに際し、吸入しないよう注意する。
臭化第二水銀 HgBr2〔特級〕
臭化第二水銀紙 臭化第二水銀5g(4.5~5.4g)にエタノール100mLを加え、穏やかに加熱して溶かし、この溶液に、クロマトグラフ用ろ紙を幅約4cm、長さ約10cmに切ったものを暗所で約1時間浸した後、試験に用いる部分に直接手を触れないようにして溶液から引き上げ、ガラス棒につり下げて自然に乾燥させる。乾燥した後、周囲を切り捨て、約20mm2に切り、さらに、四隅を切り取る。遮光して暗所に保存する。
臭化ナトリウム NaBr〔特級〕
重クロム酸カリウム K2Cr2O7〔特級〕
重クロム酸カリウム(標準試薬) K2Cr2O7〔容量分析用標準試薬〕
シュウ酸アンモニウム (NH4)2C2O4・H2O〔特級〕
シュウ酸アンモニウム試液 シュウ酸アンモニウム3.5g(3.45~3.54g)に水を加えて溶かし、100mLとする(0.25mol/L)。
シュウ酸アンモニウム試液、0.07mol/L シュウ酸アンモニウム0.8g(0.75~0.84g)を水80mLに溶かす。
シュウ酸ナトリウム(標準試薬) C2O4Na2〔容量分析用標準試薬〕
シュウ酸N-(1-ナフチル)-N′-ジエチルエチレンジアミン C18H24N2O4・1/2H2O〔N-1-ナフチル-N′-ジエチルエチレンジアミンシュウ酸塩、特級〕 遮光して保存する。
シュウ酸N-(1-ナフチル)-N′-ジエチルエチレンジアミン試液 シュウ酸N-(1-ナフチル)-N′-ジエチルエチレンジアミン1g(0.5~1.4g)に水を加えて溶かし、1,000mLとする。
臭素 Br2〔特級〕 暗赤褐色の揮発性の液体で刺激性が強く、腐食性があり、水に溶けにくく、エタノール又はエーテルに溶ける。
臭素試液 臭素を水に飽和して調製する。栓にワセリンを塗った共栓瓶に臭素2~3mLを入れ、冷水100mLを加え、密栓して振り混ぜる。遮光して、できる限り冷所に保存する。
臭素酸カリウム KBrO3〔特級〕
臭素酸カリウム・臭化カリウム試液 臭素酸カリウム1.4g(1.35~1.44g)及び臭化カリウム8.1g(8.05~8.14g)を水に溶かし、100mLとする。
酒石酸カリウムナトリウム KNaC4H4O6・4H2O〔酒石酸カリウムナトリウム(ロッセル塩(セニエット塩))、特級〕
酒石酸水素ナトリウム NaHC4H4O6・H2O〔特級〕
酒石酸水素ナトリウム試液 酒石酸水素ナトリウム1g(0.5~1.4g)に水を加えて溶かし、10mLとする(1mol/L)。用時調製する。
消化血清 黄色の粉末である。
乾燥減量 5%以下(1g,85℃,1時間)
溶解性 本品の溶液(1→100)をpH7.0に調整し、121℃で15分間高圧蒸気滅菌するとき、不溶物を認めない。
pH 本品の溶液(1→100)を121℃で15分間高圧蒸気滅菌するときのpHは、5.5~7.0でなければならない。
硝酸 HNO3〔特級、比重 約1.42〕
硝酸・塩酸試液、誘導結合プラズマ分析用 誘導結合プラズマ分析用硝酸18mL及び誘導結合プラズマ分析用塩酸2mLを量り、500mLの全量フラスコに入れ、誘導結合プラズマ分析用水を標線まで加え、蓋をし、よく振り混ぜる。
硝酸、希 硝酸10.5mLに水を加えて100mLとする(10%)。
硝酸銀 AgNO3〔特級〕
硝酸銀試液 硝酸銀17.5g(17.45~17.54g)に水を加えて溶かし、1,000mLとする(0.1mol/L)。遮光して保存する。
硝酸コバルト Co(NO3)2・6H2O〔特級〕
硝酸ストロンチウム Sr(NO3)2〔硝酸ストロンチウム(無水)、特級〕
硝酸第二水銀試液 黄色酸化第二水銀40g(39.5~40.4g)に硝酸32mL及び水15mLを加えて溶かす(2mol/L)。遮光した共栓瓶に保存する。
硝酸鉛 Pb(NO3)2〔特級〕
蒸留水 精製水を用いる。
硝酸マグネシウム Mg(NO3)2・6H2O〔特級〕
硝酸マグネシウム試液 酸化マグネシウム3.75g(3.745~3.754g)に水30mLを加え、硝酸10mLを徐々に加えて溶かす。放冷した後、水を加えて50mLとする。
硝酸、誘導結合プラズマ分析用 HNO3〔微量金属測定用、69~70%〕
食塩 NaCl〔塩化ナトリウム、白色〕
シリカゲル 無定形の一部水加性のケイ酸で、不定形ガラス状顆粒である。乾燥剤用として水分吸着により変色する変色料を含ませたものもある。110℃で乾燥して元の色に戻す。
強熱減量 6%以下(2g,950±50℃)
水分吸着能 31%以上
本品約10gを0.001gの桁まで量り、その数値を記録し、はかり瓶に入れ、蓋を除いて比量1.19の硫酸で湿度を80%とした容器内に24時間入れた後、質量を量り、試料に対する増量を求める。
シリカゲル、液体クロマトグラフ用 シリカゲルを液体クロマトグラフ用に製造した上質のポーラスシリカとする。
シリカゲル、薄層クロマトグラフ用 シリカゲルを薄層クロマトグラフ用に製造した上質のものとする。
シリカゲル(蛍光剤入り)、薄層クロマトグラフ用 薄層クロマトグラフ用シリカゲルに蛍光剤を加えたものとする。
シリコーンエマルジョン 消泡用シリコーンコンパウンドを乳化剤で水に分散させた乳白色の液体で水とよく混ざる。
pH 2~5(25℃)
蒸発残分 11.5%以上(1g,105℃,2時間)
シリコーン油 無色で、澄明の液体で、臭いはない。常温における粘度50~100センチストークスとする。
水酸化カリウム KOH〔特級〕 KOH85.0%以上を含む。
水酸化カリウム試液 水酸化カリウム6.5g(6.45~6.54g)に水を加えて溶かし、100mLとする(1mol/L)。ポリエチレン瓶に保存する。
水酸化カリウム・エタノール試液 水酸化カリウム10g(9.5~10.4g)にエタノールを加えて溶かし、100mLとする。用時調製する。
水酸化カリウム・エタノール試液、希 水酸化カリウム35g(34.5~35.4g)に水20mLを加えて溶かし、エタノールを加えて1,000mLとする(0.5mol/L)。密栓して保存する。
水酸化カルシウム Ca(OH)2〔1級〕
水酸化カルシウム試液 水酸化カルシウム3g(2.5~3.4g)に冷蒸留水1,000mLを加え、1時間ときどき強く振り混ぜた後、静置し、用時、上澄液を用いる(0.02mol/L)。
水酸化カルシウム、pH測定用 〔水酸化カルシウム、1級〕 23~27℃で得た飽和溶液の25℃におけるpHが12.45のものを用いる。
水酸化ナトリウム NaOH〔特級〕 NaOH95.0%以上を含む。
水酸化ナトリウム試液、2.5mol/L 水酸化ナトリウム107g(106.5~107.4g)に水を加えて溶かし、1,000mLとする。ポリエチレン瓶に保存する。
水酸化ナトリウム試液、2mol/L 水酸化ナトリウム86g(85.5~86.4g)に水を加えて溶かし、1,000mLとする。ポリエチレン瓶に保存する。
水酸化ナトリウム試液、1mol/L 水酸化ナトリウム4.3g(4.25~4.34g)に水を加えて溶かし、100mLとする。ポリエチレン瓶に保存する。
水酸化ナトリウム試液、0.5mol/L 水酸化ナトリウム22g(21.5~22.4g)に水を加えて溶かし、1,000mLとする。ポリエチレン瓶に保存する。
水酸化ナトリウム試液、0.2mol/L 1mol/L水酸化ナトリウム試液200mLに水を加えて1,000mLとする。ポリエチレン瓶に保存する。
水酸化ナトリウム試液、0.05mol/L 1mol/L水酸化ナトリウム試液50mLに水を加えて1,000mLとする。ポリエチレン瓶に保存する。
水酸化ナトリウム試液、希 水酸化ナトリウム4.3g(4.25~4.34g)を量り、新たに煮沸した水を加えて溶かし、1,000mLとする(0.1mol/L)。用時調製する。
水酸化ナトリウム試液、酵素力試験用 水酸化ナトリウム32.0g(31.95~32.04g)に水を加えて溶かし、300mLとする。ポリエチレン瓶に保存する。
0.1mol/L水酸化ナトリウム・メタノール試液 水酸化ナトリウム4.5g(4.45~4.54g)にメタノールを加えて溶かし、1,000mLとし、ガラスろ過器を用いてろ過する。
0.05mol/L水酸化ナトリウム・メタノール試液 0.1mol/L水酸化ナトリウム・メタノール試液50mLにメタノールを加えて100mLとする。
水酸化バリウム Ba(OH)2・8H2O〔特級〕 密栓して保存する。
水酸化リチウム LiOH〔特級〕
膵消化カゼイン カゼインをパンクレアチンを用いて酵素分解したものであり、灰黄色の粉末で、特異な臭いがある。水に溶け、淡黄色を呈する。
pH 9.5~7.0(2%水溶液)
窒素含量 10%以下
バンスライク法によるアミノ酸窒素量/総窒素量(%) 0.25~0.50
トリプトファン含量 1.5%以下(微生物学的定量法によるものとする。)
乾燥減量 7%以下
強熱残分 15%以下
水素 H2〔標準物質、3級〕
水素化ホウ素ナトリウム NaBH4 本品は、白色~灰白色の結晶、粉末又は塊である。本品は、水に溶けやすく、エーテルに極めて溶けにくい。400℃で分解され、引火性で、吸湿性がある。火気を避けて保存する。
水素化ホウ素ナトリウム試液 水素化ホウ素ナトリウム4.0g(3.95~4.04g)に水酸化ナトリウム試液を加えて溶かし、100mLとする。
スチグマステロール C29H48O〔特級〕
スルファニルアミド NH2C6H4SO2NH2〔特級〕
スルファニルアミド試液 スルファニルアミド1g(0.5~1.4g)にエタノールを加えて溶かし、100mLとし、これに0.5mol/L硫酸100mLを加えて200mLとする。用時調製する。
スルファニル酸 H2NC6H4SO3H〔特級〕
スルファニル酸試液 スルファニル酸4g(3.5~4.4g)に希硫酸を加えて250mLとする。
スルファミン酸(標準試薬) HOSO2NH2〔容量分析用標準試薬〕
スルファミン酸アンモニウム NH4OSO2NH2〔1級〕
スルファミン酸アンモニウム試液 スルファミン酸アンモニウム1g(0.5~1.4g)に水を加えて溶かし、40mLとする。
生菌剤試験用抗生物質溶液 クロラムフェニコール0.025g(0.0245~0.0254g)をメタノール1mLに溶かし、滅菌水で100mLの全量フラスコに洗い込み、これに硫酸ポリミキシンB125,000単位を加えて溶解し、滅菌水を加えて100mLとする。5℃以下で保存し、1か月以内に使用する。
精製水 精製水は、常水を蒸留して又はイオン交換樹脂を通して精製した水である。
物理的・化学的性質 本品は、無色で、澄明の液体で、臭い及び味はない。
液性 本品のpHは、5~7である。
塩化物 本品50mLに硝酸3滴及び硝酸銀試液0.5mLを加えるとき、溶液は変化しない。
硫酸塩 本品50mLに塩化バリウム試液0.5mLを加えるとき、溶液は変化しない。
アンモニウム塩 本品50mLにネスラー試液0.5mLを加えるとき、溶液は変化しない。
重金属 本品40mLに希酢酸1mL及び新たに作製した硫化水素試液10mLを加え、10分間放置するとき、その溶液の色は、本品50mLに希酢酸1mLを加えた溶液の色より濃くてはならない。
過マンガン酸カリウム還元性物質 本品100mLに希硫酸10mLを加え、煮沸し、0.02mol/L過マンガン酸カリウム試液0.1mLを加え、更に10分間煮沸するとき、試液の色は消えない。
蒸発残留物 本品100mLを水浴上で蒸発乾固し、さらに、100℃で恒量になるまで乾燥するとき、その残留物は、1mg以下である。
生理食塩液 〔日局〕
赤外用臭化カリウム 臭化カリウム、赤外用の項に定める。
赤色リトマス紙 リトマス紙、赤色の項に定める。
石油エーテル 〔特級〕
石油ベンジン 〔特級〕
セルビオース C12H22O11
比旋光度〔α〕D20=+34~+35°
重金属 10μg/g以下
水分 0.5%以下
セルロース末、薄層クロマトグラフ用 セルロース末を薄層クロマトグラフ用に製造した上質のもの。
セルロース末(蛍光剤入り)、薄層クロマトグラフ用 セルロース末、薄層クロマトグラフ用に蛍光剤を加えたもの。
大豆ペプトン 淡黄色の粉末である。
乾燥減量 5%以下(1g,85℃,1時間)
溶解性 本品の溶液(1→100)を121℃で15分間高圧蒸気滅菌するとき、不溶物を認めない。
pH 本品の溶液(1→100)を121℃で15分間高圧蒸気滅菌するときのpHは、7.0~7.5でなければならない。
多孔性スチレンジビニルベンゼン共重合体 多孔性スチレンジビニルベンゼン共重合体を液体クロマトグラフ用に製造した上質のものとする。
多孔性スチレンジビニルベンゼン共重合体精製樹脂 多孔性スチレンジビニルベンゼン共重合体を20倍量のメタノールに30分間浸漬し、攪拌した後、上澄液を捨て、順次20倍量の水、10倍量のアセトン、10倍量のメタノール・0.2mol/L塩酸試液混液(7:3)、20倍量の水及び20倍量のメタノール(1→2)で同様に処理し、メタノール(1→2)に浸漬して用いる。
脱脂綿 〔日局〕
タルトラトアンチモン(Ⅲ)酸カリウム K(SbO)C4H4O6・1/2H2O〔酒石酸アンチモニルカリウム、特級〕
タングステン酸ナトリウム Na2WO4・2H2O〔特級〕
炭酸アンモニウム 〔特級〕
炭酸アンモニウム試液 炭酸アンモニウム20g(19.5~20.4g)及びアンモニア試液20mLに水を加えて溶かし、100mLとする。
炭酸カリウム K2CO3〔炭酸カリウム(無水)、特級〕
炭酸水素ナトリウム NaHCO3〔炭酸水素ナトリウム(重炭酸ナリトウム)、特級〕
炭酸水素ナトリウム緩衝液(pH9.3)炭酸水素ナトリウム21.0g(20.95~21.04g)に水を加えて溶かし、1mol/L塩酸試液でpH9.3に調整した後、更に水を加えて500mLとする。
炭酸水素ナトリウム・水酸化ナトリウム緩衝液(pH9.0) 炭酸水素ナトリウム84.0g(83.95~84.04g)に水を加えて溶かし、900mLとする。この溶液に水酸化ナトリウム溶液(3→10)を加えてpHを9.0とし、水を加えて1,000mLとする。
炭酸水素ナトリウム、pH測定用 NaHCO3〔炭酸水素ナトリウム(重炭酸ナトリウム)、pH測定用〕
炭酸ナトリウム Na2CO3・10H2O〔特級〕
炭酸ナトリウム(標準試薬) Na2CO3〔容量分析用標準試薬〕
炭酸ナトリウム、pH測定用 Na2CO3〔炭酸ナトリウム(無水)、pH測定用〕
炭酸水素ナトリウム・メタノール試液 炭酸水素ナトリウム0.25g(0.245~0.254g)にメタノール1,000mLを加えて溶かし、ろ過する。
炭酸ナトリウム、無水 Na2CO3〔炭酸ナトリウム(無水)、特級〕
炭酸ナトリウム試液 無水炭酸ナトリウム10.5g(10.45~10.54g)に水を加えて溶かし、100mLとする(1mol/L)。
炭酸ナトリウム試液、0.55mol/L 無水炭酸ナトリウム58.3g(58.25~58.34g)に水を加えて溶かし、1,000mLとする。
炭酸マグネシウム 〔日局〕
チオグリコール酸ナトリウム HSCH2COONa〔日局〕
チオシアン酸アンモニウム NH4SCN〔特級〕
チオシアン酸アンモニウム試液 チオシアン酸アンモニウム8g(7.5~8.4g)に水を加えて溶かし、100mLとする(1mol/L)。
チオシアン酸アンモニウム・硝酸コバルト試液 チオシアン酸アンモニウム17.4g(17.35~17.44g)及び硝酸コバルト2.8g(2.75~2.84g)を水に溶かし、100mLとする。
チオシアン酸カリウム KSCN〔特級〕
チオシアン酸カリウム試液 チオシアン酸カリウム1g(0.5~1.4g)に水を加えて溶かし、10mLとする。
チオシアン酸水銀アンモニウム試液 チオシアン酸アンモニウム30g(29.5~30.4g)及び塩化第二水銀27g(26.5~27.4g)に水を加えて溶かし、1,000mLとする。
チオジエチレングリコール S(CH2CH2OH)2〔β-チオジグリコール、日局]
チオ硫酸ナトリウム Na2S2O3・5H2O〔特級〕
窒素ガス N2〔窒素、日局〕
チモール CH3C6H3(OH)CH(CH3)2〔日局〕
チモールフタレイン C28H30O4〔特級〕 変色範囲pH(無色)9.3~10.5(青)
チモールフタレイン試液 チモールフタレイン0.1g(0.05~0.14g)にエタノール100mLを加えて溶かし、必要ならば、ろ過する。
チモールブルー C27H30O5S〔特級〕 変色範囲pH酸性側(赤)1.2~2.8(黄)、アルカリ性側(黄)8.0~9.6(青)
チモールブルー試液 チモールブルー0.1g(0.05~0.14g)にエタノール100mLを加えて溶かし、必要ならば、ろ過する。
チモールブルー・ジメチルホルムアミド試液 チモールブルー0.1g(0.05~0.14g)にジメチルホルムアミド100mLを加えて溶かす。
チモール・硫酸試液 チモール0.5g(0.45~0.54g)に硫酸5mLを加えて溶かし、エタノールを加えて100mLとする。
中和エタノール エタノール、中和の項に定める。
2―デアミノ―2―ヒドロキシメチオニンイソプロピルエステル、定量用 2―デアミノ―2―ヒドロキシメチオニンイソプロピルエステル製造用原体 定量するとき、2―デアミノ―2―ヒドロキシメチオニンイソプロピルエステル(C8H16O3S)98.0%以上のものに限る。
定量用L-イソロイシン L-イソロイシン、定量用の項に定める。
定量用エトキシキン エトキシキン、定量用の項に定める。
定量用塩酸L-ヒスチジン 塩酸L-ヒスチジン、定量用の項に定める。
定量用ギ酸ナトリウム ギ酸ナトリウム、定量用の項に定める。
定量用酢酸dl -α-トコフェロール 酢酸dl -α-トコフェロール、定量用の項に定める。
定量用2―デアミノ―2―ヒドロキシメチオニンイソプロピルエステル 2―デアミノ―2―ヒドロキシメチオニンイソプロピルエステル、定量用の項に定める。
定量用ニンヒドリン試液 ニンヒドリン試液、定量用の項に定める。
定量用d -ビオチン d -ビオチン、定量用の項に定める。
定量用プロピオン酸ナトリウム プロピオン酸ナトリウム、定量用の項に定める。
定量用DL-メチオニン DL-メチオニン、定量用の項に定める。
定量用メナジオン亜硫酸水素ジメチルピリミジノール メナジオン亜硫酸水素ジメチルピリミジノール、定量用の項に定める。
定量用メナジオン亜硫酸水素ナトリウム メナジオン亜硫酸水素ナトリウム、定量用の項に定める。
DEAE-セファデックスA-25、クロマトグラフ用 水吸収度2.5のクロマトグラフ用DEAE-セファデックス
滴定用2,6-ジクロルフェノールインドフェノールナトリウム試液 2,6-ジクロルフェノールインドフェノールナトリウム試液、滴定用の項に定める。
テトラヒドロフラン C4H8O〔特級〕
テトラブチルアンモニウムヒドロキシド試液 〔日局〕
デンプン 〔特級〕
デンプン試液 デンプン1g(0.5~1.4g)を冷水10mLとよくすり混ぜ、これを熱湯200mL中に絶えずかき混ぜながら徐々に注ぎ込み、溶液が半透明となるまで煮沸し、溶液を放置した後、上澄液を用いる。用時調製する。
ドラーゲンドルフ試液 次硝酸ビスマス0.85g(0.845~0.854g)に氷酢酸10mLを加えて溶かし、水40mLを加えてA液とする。ヨウ化カリウム8g(7.5~8.4g)に水20mLを加えて溶かし、B液とする。
使用直前にA液、B液及び氷酢酸のそれぞれ等容量を混ぜて用いる。A液及びB液は、遮光して保存する。
ドラーゲンドルフ試液、噴霧用 ドラーゲンドルフ試液のA液及びB液の等容量混液4mLに酢酸(1→5)20mLを加える。用時調製する。
トリエチルアミン (C2H5)3N〔日局〕
トリクロル酢酸 CCl3COOH〔特級〕
トリクロル酢酸試液A トリクロル酢酸7.20g(7.195~7.204g)に水を加えて溶かし、100mLとする。
トリクロル酢酸試液B トリクロル酢酸1.80g(1.795~1.804g)及び無水酢酸ナトリウム1.80g(1.795~1.804g)に6mol/L酢酸試液5.5mL及び水を加えて溶かし、100mLとする。
トリフェニルクロルメタン (C6H5)3CCl〔トリフェニルクロルメタン(塩化トリチル)、特級〕
トリプシン ウシの膵臓より調製する。
消化度 本品0.01g(0.005~0.014g)に水500mLを加えて溶かし、試料溶液とする。この試料溶液2mLにカゼイン試液5mL及び水3mLを加え、混和し、40℃に1時間放置した後、酢酸・エタノール試液3滴を加えるとき、沈殿を生じない。
トリプトース カゼイン製ペプトンと獣肉製ペプトンを混合したものである。
DL-トリプトファン C11H12N2O2 白色~微褐色の結晶性の粉末である。希塩酸に溶けやすく、水に溶けにくく、エタノールに極めて溶けにくい。
溶状 本品1g(0.5~1.4g)に水20mLを加え、加温溶解するとき、溶液は、澄明である。
塩化物 0.02%以下
硫酸塩 0.03%以下
乾燥減量 0.3%以下(1g,105℃,3時間)
強熱残分 0.05%以下(1g)
窒素含量 13.4~13.8%(窒素定量法によるものとする。)
トリメチルアミン塩酸塩 (CH3)3N・HCl〔特級〕
トルエン C6H5CH3〔特級〕
トルエン、無水 C6H5CH3 トルエン500mLに無水硫酸ナトリウム10g(9.5~10.4g)を加え、12時間放置した後、硫酸ナトリウムを除く。
ο-トルエンスルホンアミド C7H9NO2S 無色の結晶又は白色の結晶性粉末である。
融点 157~160℃
純度試験 p-トルエンスルホンアミド 本品の酢酸エチル溶液(1→5,000)につき、サッカリンナトリウム製造用原体の純度試験⑥の操作条件でガスクロマトグラフ法により試験を行うとき、本品以外のピークを認めてはならない。
ナトリウム、金属 Na〔ナトリウム、特級〕
ナトリウムペンタシアノアンミンフェロエート Na3〔Fe(CN)5NH3〕・nH2O〔1級〕
N-1-ナフチル-エチレンジアミン二塩酸塩〔特級〕
α-ナフトール C10H7OH〔特級〕 遮光して保存する。
α-ナフトールベンゼイン C27H20O3〔特級〕 赤褐色の粉末で、水に溶けないが、エタノール、ベンゼン、エーテル又は氷酢酸に溶ける。酸性で黄赤色、アルカリ性で緑色を呈する。
α-ナフトールベンゼイン試液 α-ナフトールベンゼイン0.2g(0.15~0.24g)に氷酢酸を加えて溶かし、100mLとする。
溶状 本品0.1g(0.05~0.14g)をエタノール100mLに溶かすとき、溶液は、赤色で、澄明である。
鋭敏度 本品のエタノール溶液(1→1,000)0.2mLに、新たに煮沸し冷却した水100mLを加え、0.1mol/L水酸化ナトリウム溶液0.1mLを加えるとき、緑色を呈し、さらに、0.1mol/L塩酸0.2mLを加えるとき、黄赤色に変わる。
二塩化エチレン C2H2Cl2〔特級〕
肉エキス 本品は、脂肪分をほとんど含まない部分の動物肉を水で煮沸又は加温し、得られた肉汁を低温、減圧下で濃稠なペースト状となるまで濃縮したものである。本品は、下記の物理的・化学的性質及び規格に適合するものと、試験において同等以上又は試験において支障を来さない限り、類似の適当な市販品を用いることができる。
物理的・化学的性質 本品は、黄褐色~濃暗褐色のペースト状で、僅かに酸性を示し、肉様の臭い及び味がある。
溶状 本品をよくかき混ぜ、25g(24.5~25.4g)を量り、水を加えて250mLとし、よく振り混ぜ、試料溶液とするとき、溶液は、澄明又は混濁して溶けるが、沈殿物は認めない。
総固形物 試料溶液10mLを量り、磁製皿に入れ、100~110℃で16時間加熱するとき、残留物の質量は、650mg以上である。
強熱残分 上記残留物を強熱残分試験法により強熱するとき、総固形物質量の30%以下である。
食塩換算塩素 上記強熱残分で得た残分に、水約50mLを加え、振り混ぜ、100mLの全量フラスコに入れ、硝酸2~4滴及び全量ピペットを用いて0.1mol/L硝酸銀液10mLを加え、更に水を加えて100mLとし、よく振り混ぜ、ろ過し、最初のろ液10mLを除き、次のろ液50mLを量り、硫酸第二鉄アンモニウム試液1mLを加え、0.1mol/Lチオシアン酸アンモニウム溶液で滴定するとき、塩化ナトリウムの量は、総固形物質量の6%を超えない。
0.1mol/L硝酸銀溶液1mL=5.844mgNaCl
硝酸塩 試料溶液10mLを量り、活性炭1.5g(1.45~1.54g)を加え、1分間煮沸した後、水を加えて約10mLとし、ろ過する。ろ液にジフェニルアミン試液3滴を加えるとき、溶液は、青色を呈さない。
アンモニア態窒素 試料溶液100mLを量り、炭酸バリウム5g(4.5~5.4g)及び水100mLを加え、0.05mol/L硫酸50mLを入れた受器中に、蒸留液が約100mLになるまで水蒸気蒸留する。蒸留液に、メチルレッド試液1~3滴を加え、0.1mol/L水酸化ナトリウム溶液で滴定するとき、アンモニア態窒素の総量は、総固形物質量の0.35%を超えない。
0.05mol/L硫酸1mL=1.703mgNH3
ニコチン酸 C6H5NO2〔日局〕
二酸化炭素 CO2〔日局〕
p-ニトロアニリン O2NC6H4NH2〔1級〕
3―ニトロオキシプロパノール
含量 98.0%以上
定量法 本品約100mgを有効数字3桁まで量り、10mLの褐色全量フラスコに入れ、内標準溶液5mLを全量ピペットを用いて量り、加え、よく振り混ぜ、バイアルに移し、試料溶液とする。この溶液につき、3―ニトロオキシプロパノール製造用原体の定量法に定める測定条件でガスクロマトグラフ法により試験を行う。試料溶液注入後、測定時間に現れる、エタノール由来のピーク及びノナン酸メチルのピークを除いた、全ての成分のピーク面積の総和を100とし、それに対する3―ニトロオキシプロパノールのピーク面積百分率を求める。
5-ニトロソ-8-オキシキノリン C9H5NOHNO 本品は、暗灰緑色の結晶性粉末である。
融点 約245℃(分解)
溶状 本品0.1g(0.05~0.14g)を硫酸100mLに溶かすとき、その溶液は、澄明である。
鋭敏度 レゾルシンのエタノール溶液(1→1,000)0.05mLをるつぼに入れ、水浴上で蒸発乾固し、放冷した後、これに本品の硫酸溶液(1→1,000)0.05mLを加えて加熱するとき、赤紫色となる。
α-ニトロソ-β-ナフトール C10H7NO2〔特級〕
α-ニトロソ-β-ナフトール試液、強 α-ニトロソ-β-ナフトール1g(0.5~1.4g)に氷酢酸25mLを加え、加温して溶かした後、水25mLを加え、加温して溶かし、更に水25mLを加え、1時間放置した後、ろ過する。
1-ニトロソ-2-ナフトール-3,6-ジスルホン酸二ナトリウム C10H5NNa2O8S2〔1-ニトロソ-2-ナフトール-3,6-ジスルホン酸二ナトリウム(ニトロソR塩)、特級〕
ο-ニトロフェニル-β-D-ガラクトピラノシド C12H15NO8 白色の結晶性粉末で、臭いはない。水にやや溶けにくく、エタノールに溶けにくい。
融点 193~194℃
比旋光度 本品1.0gを0.01gの桁まで量り、その数値を記録し、水を加えて溶かし、100mLの全量フラスコに入れ、更に水を標線まで加えて100mLとし、この溶液につき、層長100mmで旋光度を測定するとき、〔α〕D18=-51.9℃である。
強熱残分 0.05%以下(2g)
ニトロプルジドナトリウム Na2Fe(CN)5(NO)・2H2O〔特級〕
ニトロプルシドナトリウム試液 ニトロプルシドナトリウム1g(0.5~1.4g)に水を加えて溶かし、20mLとする。用時調製する。
ニトロベンゼン C6H5NO2〔特級〕
乳酸 C3H6O3〔特級〕
乳酸塩緩衝液、0.1mol/L、酵素力試験用 乳酸9.01g(9.005~9.014g)に水を加えて溶かし、900mLとする。この溶液に希水酸化ナトリウム試液を加えて所定のpHに調整し、水を加えて1,000mLとする。
乳酸試液、1mol/L 乳酸90.1g(90.05~90.14g)に水を加えて溶かし、1,000mLとする。
乳酸試液、0.05mol/L 乳酸4.5g(4.45~4.54g)に水を加えて溶かし、1,000mLとする。
乳製カゼイン カゼイン、乳製の項に定める。
乳糖 C12H22O11・H2O〔日局〕
二硫化炭素 CS2〔特級〕
鶏卵黄液 鶏卵から卵黄のみを無菌的にとり、卵黄と同量の水で希釈する。用時調製する。
ニンヒドリン C9H4O3・H2O〔ニンヒドリン(抱水トリケトヒドリンデン)、特級〕
ニンヒドリン・アスコルビン酸試液 ニンヒドリン0.25g(0.245~0.254g)及びアスコルビン酸0.01g(0.005~0.014g)を量り、水を加えて溶かし、50mLとする。用時調製する。
ニンヒドリン、アミノ酸分析用 C9H6O4〔アミノ酸分析用〕
ニンヒドリン試液 ニンヒドリン0.2g(0.15~0.24g)に水を加えて溶かし、10mLとする。用時調製する。
ニンヒドリン試液、アミノ酸分析用 アミノ酸分析用ニンヒドリン30g(29.5~30.4g)、水酸化ホウ素ナトリウム134mg(133.5~134.4mg)、酢酸緩衝液800mL及びメチルセロソルブ1,200mLを混合し、窒素ガスを吹きつけながら溶解する。冷蔵保存する。
ニンヒドリン試液、定量用 ニンヒドリン5g(4.5~5.4g)、塩化第二銅6.7g(6.65~6.74g)、クエン酸緩衝液125mL及びメチルセロソルブ375mLに水を加えて1,000mLとする。調製した後2週間は、安定である。
ニンヒドリン・ブタノール試液 ニンヒドリン0.3g(0.25~0.34g)をn-ブタノール100mLに溶かし、氷酢酸3mLを加える。
ネスラー試液 ヨウ化カリウム10g(9.5~10.4g)に水10mLを加えて溶かし、かき混ぜながら塩化第二水銀飽和溶液を赤色沈殿が溶けずに僅かに残るまで加える。これに水酸化カリウム30g(29.5~30.4g)を水60mLに溶かした溶液を加え、さらに、塩化第二水銀飽和溶液1mL及び水を加えて200mLとする。沈殿を沈着させた上澄液を用いる。本液2mLを塩化アンモニウム溶液(1→300,000)100mLに加えるとき、30秒以内に黄褐色を呈する。
濃ヨウ化カリウム試液 ヨウ化カリウム試液、濃の項に定める。
ノナン酸メチル CH3(CH2)7COOCH3 99.8%以上
L-ノルロイシン C6H13NO2〔アミノ酸分析用〕
L-ノルロイシン試液 L-ノルロイシン16mg(15.5~16.4mg)に1mol/L塩酸を加えて溶かし、50mLとする。
ハイドロサルファイトナトリウム Na2S2O4〔ハイドロサルファイトナトリウム(ハイドロサルファイト)、1級〕
薄層クロマトグラフ用シリカゲル シリカゲル、薄層クロマトグラフ用の項に定める。
薄層クロマトグラフ用シリカゲル(蛍光剤入り) シリカゲル(蛍光剤入り)、薄層クロマトグラフ用の項に定める。
薄層クロマトグラフ用セルロース末(蛍光剤入り) セルロース末(蛍光剤入り)、薄層クロマトグラフ用の項に定める。
バナジン酸アンモニウム NH4VO3〔特級〕
バニリン C6H3CHO(OCH3)(OH) 白色の結晶で、特異な臭い及び味を有する。
融点 81~83℃
乾燥減量 1.0%以下(1g,シリカゲル,4時間)
強熱残分 0.05%以下(1g)
遮光して保存する。
バニリン・塩酸試液 バニリン10mg(9.5~10.4mg)にエタノール1mLを加えて溶かし、水1mL及び塩酸6mLを加える。用時調製する。
バニリン・硫酸・エタノール試液 バニリン3g(2.5~3.4g)に無水エタノールを加えて溶かし、100mLとした溶液に、硫酸0.5mLを加える。
バニリン・硫酸・エタノール発色試液 バニリン3g(2.5~3.4g)に無水エタノールを加えて溶かし、100mLとした溶液に、硫酸3mLを加える。
パパイン消化大豆 大豆たん白をパパインを用いて酵素分解したものである。適当な市販品を用いる。
パパイン消化肝臓 雄牛の肝臓をパパインを用いて酵素分解したものである。適当な市販品を用いる。
パラオキシ安息香酸第二ブチル HOC6H4CO2CH2CH(CH3)2 白色~淡黄白色の結晶性の粉末で、臭いはない、又は僅かに特異な臭いがある。
溶状 本品0.5g(0.45~0.54g)をアセトニトリル10mLに溶かすとき、溶液は、無色~淡黄色で、澄明で、異物を認めない。
融点 59~61℃
パラオキシ安息香酸ブチル HOC6H4CO2CH2CH2CH2CH3〔日局〕
パラクレゾール C6H4(OH)CH3〔特級〕
パラフィン、流動 〔軽質流動パラフィン、日局〕
バルビタール緩衝液 バルビタールナトリウム15g(14.5~15.4g)に水700mLを加えて溶かし、希塩酸を加えてpH7.6とした後ろ過する。
バルビタールナトリウム C8H11N2NaO3 白色の結晶又は結晶性の粉末で、臭いはなく、味は苦い。水に溶けやすく、エタノールに溶けにくく、エーテルにほとんど溶けない。
pH 本品の水溶液(1→200)のpHは、9.9~10.3である。
乾燥減量 1.0%以下(1g,105℃,4時間)
含量 98.5%以上
定量法 本品を105℃で4時間乾燥し、その約0.5gを0.001gの桁まで量り、その数値を記録し、分液漏斗に入れ、水20mLを加えて溶かし、エタノール5mL、希塩酸10mLを加え、クロロホルム50mLで抽出する。更にクロロホルム25mLで3回抽出し、全クロロホルム抽出液を合わせ、水5mLずつで2回洗い、洗液はクロロホルム10mLずつで2回抽出し、前後のクロロホルム抽出液を合わせ、三角フラスコ中にろ過する。ろ紙をクロロホルム5mLずつで3回洗い、ろ液及び洗液を合わせ、エタノール10mLを加え、0.1mol/L水酸化カリウム・エタノール溶液で滴定する(指示薬 アリザリンエローGG・チモールフタレイン試液2mL)。この場合において、滴定の終点は、溶液の黄色が淡青色を経て紫色に変わるときとする。同様の方法で空試験を行い補正する。
0.1mol/L水酸化カリウム・エタノール溶液1mL=20.62mgC8H11N2NaO3
バレイショデンプン〔日局〕
パントテン酸カルシウム Ca(C9H16NO3)2〔日局〕
pH測定用四シュウ酸カリウム 四シュウ酸カリウム、pH測定用の項に定める。
pH測定用水酸化カルシウム 水酸化カルシウム、pH測定用の項に定める。
pH測定用炭酸水素ナトリウム 炭酸水素ナトリウム、pH測定用の項に定める。
pH測定用炭酸ナトリウム 炭酸ナトリウム、pH測定用の項に定める。
pH測定用フタル酸水素カリウム フタル酸水素カリウム、pH測定用の項に定める。
pH測定用ホウ酸ナトリウム ホウ酸ナトリウム、pH測定用の項に定める。
pH測定用無水リン酸一水素ナトリウム リン酸一水素ナトリウム、無水、pH測定用の項に定める。
pH測定用リン酸二水素カリウム リン酸二水素カリウム、pH測定用の項に定める。
d-ビオチン、定量用 d -ビオチン製造用原体。ただし、定量するとき、d -ビオチン(C10H16N2O3S)99.0%以上のものに限る。
ヒ化水素吸収液 ジエチルジチオカルバミン酸銀0.50g(0.495~0.504g)をピリジンに溶かし、100mLとする。この溶液は、遮光した共栓瓶に入れ、冷所に保存する。
ピクリン酸 HOC6H2(NO2)3〔2,4,6-トリニトロフェノール(ピクリン酸)、特級〕 密閉し、火気を避けて冷所に保存する。
ピクリン酸試液 ピクリン酸1g(0.5~1.4g)に熱湯100mLを加えて完全に溶解した後、冷却し、必要ならば、ろ過する。
ピクリン酸・トルエン試液 ピクリン酸0.20g(0.195~0.204g)に無水トルエンを加えて溶かし、1,000mLとする。
ピクリン酸ナトリウム (NO2)3C6H2ONa・H2O〔特級〕
ピクリン酸ナトリウム試液 ピクリン酸ナトリウム0.4g(0.35~0.44g)及び水酸化ナトリウム4g(3.5~4.4g)に水を加えて溶かし、1,000mLとする。
ヒ酸二ナトリウム Na2HAsO4・7H2O〔特級〕
ヒ酸二ナトリウム試液 ヒ酸二ナトリウム6.0g(5.95~6.04g)に水を加えて溶かし、50mLとする。
非水滴定用酢酸第二水銀試液 酢酸第二水銀試液、非水滴定用の項に定める。
非水滴定用氷酢酸 酢酸、氷、非水滴定用の項に定める。
ヒ素モリブデン酸試液 モリブデン酸アンモニウム50g(49.5~50.4g)を量り、水900mLを加え、加温して溶かし、放冷した後、硫酸42mLを全量ピペットを用いて加え、さらに、ヒ酸二ナトリウム試液50mLを加えた後、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとし、37℃で一昼夜放置する。
25-ヒドロキシコレカルシフェロール C27H44O2・H2O97.0%以上を含む。
ヒドロキシセンデュラマイシンナトリウム C45H75O17Na 白色の結晶性の粉末である。
純度試験 本品1g(0.5~1.4g)を量り、メタノール1mLを加えて溶かし、この溶液5μLを薄層クロマトグラフ用シリカゲルを用いて調製した薄層板にスポットする。次に、酢酸エチル・氷酢酸混液(4:1)を展開溶媒として、約10cm展開した後、薄層板を乾燥する。これにバニリン・硫酸・エタノール試液を噴霧した後、105℃で10分間加熱するとき、Rf値約0.5の位置に赤褐色の単一のスポットを認め、その他のスポットを認めてはならない。
2-ヒドロキシ-m-トルイル酸 CH3C6H3(OH)COOH 白色~微赤色の結晶性の粉末である。エタノールに溶けやすく、水に溶けにくい。
融点 163~170℃
氷酢酸 酢酸、氷の項に定める。
氷酢酸、非水滴定用 酢酸、氷、非水滴定用の項に定める。
ビスマス酸ナトリウム NaBiO3〔特級〕
ビフェニル C12H10 白色~ほとんど白色の結晶~結晶性小片又は粉末である。
含量 98.0%以上
ピリジン C5H5N〔特級〕
ピリジン、カールフィッシャー用 一般試験法の水分定量法に定めるところによるものとする。
ピリジン、無水 C5H5N ピリジン100mLに水酸化ナトリウム10g(9.5~10.4g)を加え、24時間放置した後、上澄液を傾斜してとり、蒸留する。
ピロール C4H5N〔特級〕
ピロアンチモン酸カリウム K2H2Sb2O7・4H2O〔1級〕
ピロアンチモン酸カリウム試液 ピロアンチモン酸2g(1.5~2.4g)に水100mLを加え、約5分間煮沸した後、30秒以内に冷却し、これに水酸化カリウム溶液(3→20)10mLを加え、1日間放置してろ過する。
ピロガロール C6H3(OH)3〔特級〕
フィチン酸ナトリウム C6H6O24P6Na12・nH2O〔含量90%以上〕
ο-フェナントロリン C12H8N2・H2O〔特級〕
ο-フェナントロリン試液 o-フェナントロリン試液 o-フェナントロリン0.15g(0.145~0.154g)に新たに調製した硫酸第一鉄溶液(37→2,500)10mL及び希硫酸1mLを加えて溶かす。密栓して保存する。
フェニルヒドラジン C6H5NHNH2〔特級〕
フェノール C6H5OH〔日局〕
フェノールフタレイン C20H14O4〔特級〕 変色範囲pH(無色)8.3~10.0(紅色)
フェノールフタレイン試液 フェノールフタレイン1g(0.5~1.4g)にエタノール100mLを加えて溶かす。
フェリシアン化カリウム K3Fe(CN)6〔フェリシアン化カリウム(赤血カリ)、特級〕
フェリシアン化カリウム試液 フェリシアン化カリウム1g(0.5~1.4g)に水を加えて溶かし、10mLとする(0.33mol/L)。用時調製する。
フェーリング試液
銅液:硫酸銅34.66g(34.655~34.664g)に水を加えて溶かし、500mLとする。共栓瓶にほとんど全満して保存する。
アルカリ性酒石酸塩液:酒石酸カリウムナトリウム173g(172.5~173.4g)及び水酸化ナトリウム50g(49.5~50.4g)をとり、水に溶かし、500mLとする。ポリエチレン瓶に保存する。用時両液の等容量を混和する。
フェロシアン化カリウム K4Fe(CN)6・3H2O〔フェロシアン化カリウム(黄血カリ)、特級〕
フェロシアン化カリウム試液 フェロシアン化カリウム1g(0.5~1.4g)に水を加えて溶かし、10mLとする(0.25mol/L)。用時調製する。
フォリン試液 タングステン酸ナトリウム20g(19.5~20.4g)、モリブデン酸ナトリウム5g(4.5~5.4g)及び水約140mLを300mLのフラスコに入れ、これにリン酸(17→20)10mL及び塩酸20mLを加え、すり合わせの還流冷却器を付け、10時間緩やかに煮沸する。次に、硫酸リチウム30mLと水10mLを加え、さらに、臭素ごく少量を加えて濃緑色の溶液を黄色とし、冷却器を付けず15分間煮沸して過量の臭素を除く。放冷した後、水を加えて200mLとし、ガラスろ過器でろ過し、塵が混入しないようにして保存する。この溶液を原液とし、使用するとき、所定の濃度に水で薄める。
n-ブタノール CH3(CH2)2CH2OH〔n-ブチルアルコール(n-ブタノール)、特級〕
ブタノール、イソ (CH3)2CHCH2OH〔イソブチルアルコール(イソブタノール)、特級〕
ブタノール、第二 CH3CH2CH(OH)CH3〔第二ブチルアルコール、1級〕
o-フタルアルデヒド C6H4(CHO)2 本品は、淡黄~黄色の結晶である。
純度試験 類縁物質 本品1gをエタノール10mLに溶かし、検液とする。検液1mLを正確に量り、エタノールを加えて正確に100mLとし、比較液とする。検液及び比較液をそれぞれ10μLずつ量り、次の操作条件でガスクロマトグラフィーを行い、ピーク面積を測定するとき、検液の主ピーク以外のピークの合計面積は、比較液の主ピーク面積より大きくない。ただし、面積測定の範囲は、溶媒ピークの後ろから主ピークの保持時間の7倍までとする。
操作条件
検出器:熱伝導度検出器
カラム充填剤:
液相 担体に対して10%のメチルシリコーンポリマー
担体 酸及びシラン処理した177~250μmのガスクロマトグラフィー用ケイソウ土
カラム管:内径3mm、長さ2mのガラス管
カラム温度:180℃付近の一定温度
キャリヤーガス:ヘリウム
流量:毎分約50mLの一定量でo-フタルアルデヒドの保持時間が3~4分になるように調整する。
フタル酸水素カリウム(標準試薬) C6H4(COOK)(COOH)〔容量分析用標準試薬〕
フタル酸水素カリウム、pH測定用 C6H4(COOK)(COOH)〔pH測定用〕
フッカーの染色液 0.037mol/Lの塩化メチルロザニリン・エタノール試液及び0.07mol/Lのシュウ酸アンモニウム試液を混和し、一夜放置した後、ろ過する。遮光した容器に保存する。
フッ化水素酸 HF〔特級〕 HF46.0%以上を含む。
フッ化ナトリウム NaF〔特級〕
フッ化ナトリウム(標準試薬) NaF〔容量分析用標準試薬〕
ブドウ糖 C6H12O6〔日局〕
フルオレセイン C20H12O5〔特級〕
フルオレセインイソチオシアナート(アイソマーⅠ) C21H11NO5S 黄橙色の粉末である。
フルオレセイン・エタノール試液 フルオレセイン50mg(49.5~50.4mg)にエタノールを加えて溶かし、100mLとする。
フルオレセイン標識ペプチドグリカン試液、0.5mg/mL Micrococcus lysodeikticus由来のペプチドグリカンを以下の操作により蛍光物質であるフルオレセインイソチオシアナート(アイソマーI)で標識したもの。
ペプチドグリカン100mg(99.5~100.4mg)を量り、炭酸水素ナトリウム緩衝液(pH9.3)35mLを加え、よく振り混ぜて懸濁液とする。この懸濁液をフルオレセインイソチオシアナート(アイソマーI)800mg(799.5~800.4mg)に加え、更に懸濁液の入っていた容器を炭酸水素ナトリウム緩衝液(pH9.3)10mLで洗浄した後、フルオレセインイソチオシアナート(アイソマーI)に加える。毎分700回転、37℃で4時間振り混ぜた後、1,500×gで20分間遠心分離し、上澄液を捨てる。次に、この残留物に炭酸水素ナトリウム緩衝液(pH9.3)35mLを加えてよく振り混ぜた後、1,500×gで20分間遠心分離し、上澄液を捨てる。この操作を更に1回繰り返す。次に、この残留物に水35mLを加えてよく振り混ぜた後、1,500×gで20分間遠心分離し、上澄液を捨てる。この操作を更に1回繰り返す。次に、この残留物にアセトン35mLを加えてよく振り混ぜた後、1,500×gで20分間遠心分離し、上澄液を捨てる。この操作を更に1回繰り返す。さらに、この残留物にエタノール35mLを加えてよく振り混ぜた後、1,500×gで20分間遠心分離し、上澄液を捨てる。この操作を更に1回繰り返した後、凍結乾燥し、-20℃で保存する。
ブルーテトラゾリウム C40H32Cl2N8O2〔3,3′-ジアニソール-ビス〔4,4′-(3,5-ジフェニル)テトラゾリウムクロリド〕〕 淡黄色の結晶で、メタノール、エタノール及びクロロホルムに溶けやすく、水に溶けにくく、アセトン及びエーテルにほとんど溶けない。
融点 約245℃(分解)
分子吸光係数 60,000以上(252nm,メタノール溶液)
ブルーテトラゾリウム試液、アルカリ性 ブルーテトラゾリウムのメタノール溶液(1→200)1容量に、水酸化ナトリウムのメタノール溶液(3→25)3容量を加える。用時調製する。
プロテオーゼペプトン 淡黄色の粉末又は粒子である。
乾燥減量 3%以下(1g,85℃,1時間)
溶解性 本品の溶液(1→100)を121℃で15分間高圧蒸気滅菌するとき、不溶物を認めない。
pH 本品の溶液(1→100)を121℃で15分間高圧蒸気滅菌するときのpHは、6.5~7.5でなければならない。
n‐プロパノール CH3CH2CH2OH〔n‐プロピルアルコール(n‐プロパノール)、特級〕
プロパノール、イソ (CH3)2CHOH〔イソプロピルアルコール(イソプロパノール)、特級〕
プロピオン酸ナトリウム、定量用 C2H5COONa 白色の結晶、結晶性粉末又は顆粒で、臭いはない、又は僅かに特異な臭いを有する。
溶状 本品1.0g(0.5~1.4g)に水20mLを加えて溶かしたとき、溶液は、無色で、微濁である。
乾燥減量 0.5%以下(1g,110℃,3時間)
含量 99.0%以上
プロピレングリコール CH3CH(OH)CH2OH〔特級〕
ブロムクレゾールグリーン C21H14Br4O5S〔特級〕 変色範囲pH(黄)3.8~5.4(青)
ブロムクレゾールグリーン試液 ブロムクレゾールグリーン0.05g(0.045~0.054g)にエタノール100mLを加えて溶かす。必要ならば、ろ過する。
ブロムクレゾールグリーン・メチルオレンジ試液 ブロムクレゾールグリーンのエタノール溶液(1→1,000)80mLにメチルオレンジのエタノール溶液(1→1,000)20mLを加える。
ブロムクレゾールグリーン・メチルレッド試液 ブロムクレゾールグリーン0.15g(0.145~0.154g)及びメチルレッド0.1g(0.05~0.14g)をとり、無水エタノール180mLを加えて溶かし、水を加えて200mLとする。
ブロムクレゾールパープル試液 ブロムクレゾールパープル1.6g(1.55~1.64g)をエタノール100mLに溶かし、2mol/L水酸化ナトリウム試液でpH7.5(青紫色)に調整する。
N-ブロムコハク酸イミド (CH2CO)2NBr〔特級〕 白色の結晶粉末でアセトンにやや溶けやすく、水及び氷酢酸に溶けにくく、四塩化炭素に極めて溶けにくい。
融点 約175℃
ブロムチモールブルー C27H28Br2O5S〔特級〕 変色範囲pH(黄)6.0~7.6(青)
ブロムチモールブルー試液 ブロムチモールブルー試液 ブロムチモールブルー0.1g(0.05~0.14g)に希エタノール100mLを加えて溶かす。必要ならば、ろ過する。
ブロムチモールブルー・炭酸ナトリウム試液 ブロムチモールブルー0.15g(0.145~0.154g)及び無水炭酸ナトリウム0.15g(0.145~0.154g)に水を加えて溶かし、100mLとする。
ブロムフェノールブルー C19H10Br4O5S〔特級〕 変色範囲pH(黄)3.0~4.6(青紫)
ブロムフェノールブルー試液 ブロムフェノールブルー0.1g(0.05~0.14g)に希エタノール100mLを加えて溶かす。必要ならば、ろ過する。
フロログルシン C6H3(OH)3・2H2O〔特級〕
フロログルシン塩酸試液 フロログルシン0.1g(0.05~0.14g)をエタノール1mLに溶かし、塩酸9mLを加え、よくかき混ぜる。暗所に保存する。
噴霧用ドラーゲンドルフ試液 ドラーゲンドルフ試液、噴霧用の項に定める。
ヘキサン C6H14〔n‐ヘキサン、特級〕 ビタミンD定量に用いるn-ヘキサンは、一般試験法のビタミンD定量法に定めるところによるものとする。
n-ヘキサン、吸収スペクトル用〔n-ヘキサン、特級〕 水を対照液とし、吸光度を測定するとき、波長220nmで0.10以下、260nmで0.02以下であり、かつ、波長260~350nmにおいて、特異な吸収を認めないものとする。
ペクチン 植物体の非木質化組織に特有の酸性多糖類で、ペクチン酸の一部がメチルエステル化された構造をしており、無色、無臭及び無味の非晶質性物質である。5℃以下で保存する。
ペプチドグリカン 細菌などの細胞壁に存在する多糖類である。ただし、Micrococcus lysodeikticus由来のものを用いる。
ペプトン、カゼイン製 灰黄色の粉末で、特異な臭いがあるが腐敗臭はない。水に溶けるが、エタノール及びエーテルに溶けない。
消化度
① 本品1g(0.5~1.4g)に水10mLを加えて溶かし、試料溶液とする。この試料溶液1mLに、希エタノール10mLに氷酢酸1mLを加えた溶液0.5mLを層積するとき、界面に輪帯又は沈殿を生じない。また、この溶液を振り混ぜるとき、混濁しない。
② ①の試料溶液1mLに硫酸亜鉛飽和溶液4mLを加えるとき、少量の沈殿(プロテオース)を生じる。
③ ①の混液をろ過し、ろ液1mLに水3mL及び臭素試液4滴を加えるとき、赤紫色を呈する。
乾燥減量 7.0%以下(0.5g,105℃,恒量)
強熱残分 15.0%以下(0.5g)
窒素含量 10.0%以上(105℃で恒量に乾燥した後、窒素定量法によるものとする。)
ペプトン、獣肉製 本品は、帯赤黄色~褐色の粉末で、特異な臭いがあるが腐敗臭はない。水に溶けて黄褐色の弱酸性の溶液となる。エタノール及びエーテルには溶けない。
窒素含量 14.0~16.5%
乾燥減量 7%以下
強熱残分 5%以下(0.5g)
凝固性たん白質 本品の水溶液(1→70)を沸騰するまで加熱するとき、沈殿を生じない。
プロテオース 本品の水溶液(1→10)5mLに硫酸亜鉛液(50→35)20mLを加えるとき、僅かに軽い析出物が生じる。
ぺルオキソ二硫酸カリウム K2S2O8〔特級〕
ベルトラン試液A 硫酸銅のきれいな結晶40g(39.5~40.4g)を水に溶かし、1,000mLとする。共栓瓶にほとんど全満して保存する。
ベルトラン試液B 酒石酸カリウムナトリウム200g(199.5~200.4g)及び水酸化ナトリウム150g(149.5~150.4g)を水に溶かし、1,000mLとする。ゴム栓をして保存する。
ベルトラン試液C 硫酸第二鉄(過マンガン酸カリウム液を還元してはならない。)50g(49.5~50.4g)を適量の水に溶かし、硫酸200mLを加え、更に水を加えて1,000mLとする。
ベルトラン試液D 過マンガン酸カリウム5g(4.5~5.4g)を水に溶かし、1,000mLとする。
標定 シュウ酸アンモニウム0.25g(0.245~0.254g)を水100mLに溶かし、硫酸2mLを加え、60~70℃に加温した後、過マンガン酸カリウム溶液を滴定する。滴定量をamLとすれば、本液1mLは、Cu 0.2238/a gに相当する。
ベンゼン C6H6〔特級〕 ビタミンD定量に用いるベンゼンは、一般試験法のビタミンD定量法に定めるところによるものとする。
ベンゼン、残留農薬試験用
6-(8,11-ペンタデカジエニル)サリチル酸 C22H32O3
含量 75%以上
定量法 本品約5mgを0.1mgの桁まで量り、その数値を記録し、5mLの全量フラスコに入れ、アセトニトリルを加えて溶かし、標線までアセトニトリルを加えて5mLとし、試料溶液とする。この溶液20μLにつき、次の操作条件で液体クロマトグラフ法により試験を行う。試料溶液注入後、0~25分の間に現れる全ての成分のピーク面積の総和を100とし、それに対する主ピークの面積百分率を求め、含量とする。
操作条件
検出器:荷電化粒子検出器
カラム:内径4.6mm、長さ150mmのステンレス管に粒径5μmの液体クロマトグラフ用オクタデシルシリル化シリカゲルを充填する。
カラム温度:25℃付近の一定温度
移動相:液体クロマトグラフ用アセトニトリル・水・酢酸混液(80:20:1)
流量:毎分約2.0mL
6-(8,11,14-ペンタデカトリエニル)サリチル酸 C22H30O3
含量 85%以上
定量法 6-(8,11-ペンタデカジエニル)サリチル酸の定量法を準用する。
6-(8-ペンタデセニル)サリチル酸 C22H34O3
含量 80%以上
定量法 6-(8,11-ペンタデカジエニル)サリチル酸の定量法を準用する。
ホウ酸 H3BO3〔特級〕
ホウ酸ナトリウム Na2B4O7・10H2O〔ホウ酸ナトリウム(ホウ砂)、特級〕
ホウ酸ナトリウム、pH測定用〔ホウ酸ナトリウム(ホウ砂)、pH測定用〕
ホウ砂 ホウ酸ナトリウムの項に定める。
ポリオキシエチレン(23)ラウリルエーテル (C2H4O)23C12H26O
ポリソルベート20 C58H114O26 ソルビトール及び無水ソルビトールの水酸基の一部を主としてラウリン酸でエステル化し、エチレンオキシド約20分子を縮合させたものである。微黄色~黄色の液体で僅かに特異臭を有する。比重:約1.1 粘度(25℃):300~500mPa・S
ポリソルベート20試液 5mLのポリソルベート20に水を加えて溶かし、50mLとする。
ポリソルベート80 〔日局〕
ポリビニルアルコール (-CH2CHOH-)n 白色~微黄色の小粒又は粉末で、水に溶け、熱水には溶けにくい。エタノール及びエチルエーテルにほとんど溶けない。
溶状 本品0.5g(0.45~0.54g)に水100mLを加え、よくかき混ぜるとき、溶液は、澄明である。
けん化度 78~82mol%
本品約0.5gを0.001gの桁まで量り、その数値を記録し、300mLの共栓三角フラスコに入れ、水100mLを加え、12時間以上放置して溶かす。この溶液に0.2mol/L水酸化ナトリウム試液25mLを全量ピペットを用いて加え、常温で2時間放置する。次に、0.1mol/L硫酸試液25mLを全量ピペットを用いて加え、よくかき混ぜた後、0.1mol/L水酸化ナトリウム溶液で滴定する(指示薬 フェノールフタレイン試液2滴)。この場合において、滴定の終点は、溶液の赤色が消えたときとし、その滴定量をamLとする。同様の方法で空試験を行い、その滴定量をbmLとする。
けん化度(mol%)=100-(44.05A/(60.05-0.42A))
A(%)=(0.6005×(a-b)f)/試料の量(g)
f:0.1mol/L水酸化ナトリウム溶液のモル濃度係数
ポリビニルアルコール試液 ポリビニルアルコール20.0g(19.95~20.04g)を約800mLの水に懸濁し、かき混ぜながら75~80℃で約1時間加熱する。放冷した後、必要ならばろ過し、水を加えて1,000mLとする。
ホルマリン ホルムアルデヒド液の項に定める。
ホルマリン・炭酸マグネシウム試液 ホルマリンに炭酸マグネシウムを加え、振り混ぜ、飽和させた後、ろ過し、水で4倍容量とする。
ホルムアルデヒド液 HCHO〔特級〕
マイヤー試液 塩化第二水銀1.358g(1.3575~1.3584g)に水60mLを加えて溶かす。別に、ヨウ化カリウム5g(4.5~5.4g)に水10mLを加えて溶かす。両液を混和し、水を加えて100mLとする。
マグネシア試液 塩化マグネシウム5.5g(5.45~5.54g)及び塩化アンモニウム7g(6.5~7.4g)に水65mLを加えて溶かし、アンモニア試液35mLを加え、密栓した瓶に入れ、数日間放置してろ過する。溶液が澄明でないときは、使用前にろ過する。
マグネシウム末 Mg〔特級〕
マッキルベイン緩衝液、酵素力試験用 クエン酸21.02g(21.015~21.024g)に水を加えて溶かし、1,000mLとする。その溶液にリン酸一水素ナトリウム試液を加えて所定のpHに調整する。
マラカイトグリーン C52H54N4O12〔日局〕
マレイン酸 (CHCOOH)2〔特級〕
水、誘導結合プラズマ分析用 H2O〔日局〕
無アルデヒドエタノール エタノール、無アルデヒドの項に定める。ビタミンD定量用の無アルデヒドエタノールは、一般試験法のビタミンD定量法に定めるところによるものとする。
無水エタノール エタノール、無水の項に定める。
無水クエン酸 クエン酸、無水の項に定める。
無水ケイ酸 SiO2〔特級〕
無水酢酸 (CH3CO)2O〔特級〕
無水酢酸ナトリウム 酢酸ナトリウム、無水の項に定める。
無水酢酸・ピリジン試液 無水酢酸25g(24.5~25.4g)に無水ピリジンを加えて100mLとする。用時調製する。
無水炭酸カリウム 炭酸カリウム、無水の項に定める。
無水炭酸ナトリウム 炭酸ナトリウム、無水の項に定める。
無水トルエン トルエン、無水の項に定める。
無水ピリジン ピリジン、無水の項に定める。
無水硫酸銅 硫酸銅、無水の項に定める。
無水硫酸ナトリウム 硫酸ナトリウム、無水の項に定める。
無水リン酸一水素ナトリウム リン酸一水素ナトリウム、無水の項に定める。
無ヒ素亜鉛 亜鉛、無ヒ素の項に定める。
ムレキサイド色素 C8H8N6O6 金属性光沢を有する赤紫色の結晶で、吸収の極大波長は、520nmである。水中で深紫色を呈し、アルカリ溶液で深青色を呈する。
ムレキサイド指示薬 ムレキサイド色素0.2~0.4gを硫酸カリウム〔特級〕100g(99.5~100.4g)と粉砕混合する。
メタ過ヨウ素酸ナトリウム NaIO4〔特級〕
メタノール CH3OH〔メチルアルコール(メタノール)、特級〕
メタノール、カールフィッシャー用 一般試験法の水分定量法に定めるところによるものとする。
メタノール製5%水酸化ナトリウム試液 水酸化ナトリウム5g(4.5~5.4g)を水5mLに溶かし、メタノールを加えて100mLとする。放置した後、上澄液を用いる。
メタリン酸 HPO3〔特級〕
メタリン酸・酢酸試液 メタリン酸15g(14.5~15.4g)及び氷酢酸40mLに水を加えて溶かし、500mLとする。冷所に保存し、2日以内に使用する。
DL-メチオニン、定量用 DL-メチオニン、定量用 DL-メチオニン製造用原体。ただし、乾燥した後定量するとき、DL-メチオニン(C5H11NO2S)99%以上のものに限る。
メチルイソブチルケトン CH3COCH2CH(CH3)2〔特級〕
メチルエチルケトン CH3COC2H5〔特級〕
メチルオレンジ C14H14N3NaO3S〔特級〕 変色範囲pH(赤)3.1~4.4(橙黄)
メチルオレンジ試液 メチルオレンジ0.1g(0.05~0.14g)に水100mLを加えて溶かす。必要ならば、ろ過する。
メチルオレンジ・キシレンシアノールFF試液 メチルオレンジ1g(0.5~1.4g)及びキシレンシアノールFF1.4g(1.35~1.44g)に希エタノール500mLを加えて溶かす。
メチルセロソルブ エチレングリコールモノメチルエーテルの項に定める。
2-メチルビフェニル C13H12 無色~微黄色の液体である。
含量 95.0%以上
3-メチルビフェニル C13H12 無色~微黄色の液体である。
含量 95.0%以上
4-メチルビフェニル C13H12 白色~黄赤色又は緑色の粉末又は結晶である。
含量 97.5%以上
メチルレッド C15H15N3O2〔特級〕 変色範囲pH(赤)4.2~6.2(黄)
メチルレッド試液 メチルレッド0.1g(0.05~0.14g)にエタノール100mLを加えて溶かす。必要ならば、ろ過する。
メチレンブルー C15H18ClN3S・nH2O 〔メチレンブルー(2水塩、3水塩、4水塩)、特級〕
メナジオン亜硫酸水素ジメチルピリミジノール、定量用 メナジオン亜硫酸水素ジメチルピリミジノール製造用原体。ただし、定量するとき、換算した脱水物に対しメナジオン亜硫酸水素ジメチルピリミジノール(C17H18N2O6S)94.5%以上のものに限る。
メナジオン亜硫酸水素ナトリウム、定量用 メナジオン亜硫酸水素ナトリウム製造用原体。ただし、定量するとき、換算した脱水物に対しメナジオン亜硫酸水素ナトリウム(C11H8O2・NaHSO3)93.5%以上のものに限る。
メリビオース C12H22O11・H2O
比旋光度〔α〕D20=+141.2~+141.8°
水分 3.5~5.5%
2-メルカプトエタノール HSCH2CH2OH 本品は、無色澄明の液体である。
比重 d420 =1.112~1.117
メルチトース C18H32O16・H2O
溶状 本品1g(0.5~1.4g)に水20mLを加えて溶かすとき、溶液は、僅かに微濁する、又はほとんど澄明である。
強熱残分 0.2%以下
水分 2.5~4.5%
フェーリング溶液還元物質 本品0.5g(0.45~0.54g)に水10mLを加えて溶かし、さらに、フェーリング試液5mLを加え、3分間煮沸する。30分間放置したとき、溶液の色は変化しない。
メンブランフィルター(0.45μm) 繊維素誘導体よりなる多孔性フィルム状のろ紙で、ろ孔0.45μmのものを用いる。
メンブランフィルター(0.8μm) 繊維素誘導体よりなる多孔性フィルム状のろ紙で、ろ孔0.8μmのものを用いる。
モリブデン酸アンモニウム (NH4)6Mo7O24・4H2O〔特級〕
モリブデン酸アンモニウム試液 モリブデン酸アンモニウム21.2g(21.15~21.24g)に水を加えて溶かし、200mLとする(10%)。用時調製する。
モリブデン酸アンモニウム・タルトラトアンチモン(Ⅲ)酸カリウム・アスコルビン酸試液 モリブデン酸アンモニウム6g(5.5~6.4g)及びタルトラトアンチモン(Ⅲ)酸カリウム0.24g(0.235~0.244g)を水約300mLに溶解し、硫酸(2→3)120mLを加えた後、水を加えて1,000mLとする。別に、アスコルビン酸14.4g(14.35~14.44g)を水に溶かし、200mLとし、両液を混和する。用時調製する。
モリブデン酸ナトリウム Na2MoO4・2H2O〔特級〕
ヨウ化亜鉛デンプン紙 新たに調製したヨウ化亜鉛デンプン試液に定量分析用ろ紙を浸し、清浄な室で乾燥して調製する。共栓瓶に入れ、光及び湿気を避けて保存する。
ヨウ化亜鉛デンプン試液 水100mLを加熱して煮沸し、これにヨウ化カリウム0.75g(0.745~0.754g)を水5mLに溶かした溶液及び塩化亜鉛2g(1.5~2.4g)を水10mLに溶かした溶液を加え、溶液が沸騰している間にデンプン5g(4.5~5.4g)を水30mLに均質に懸濁した溶液をかき混ぜながら加え、2分間煮沸した後、冷却する。密栓して冷所に保存する。
感度 0.1mol/L亜硝酸ナトリウム溶液1mL、水500mL及び塩酸10mLの混液に浸したガラス棒を本液に接するとき、明らかに青色を呈する。
ヨウ化カリウム KI〔特級〕
ヨウ化カリウム試液 ヨウ化カリウム16.5g(16.45~16.54g)に水を加えて溶かし、100mLとする(1mol/L)。遮光して保存する。用時調製する。
ヨウ化カリウム試液、濃 ヨウ化カリウム30g(29.5~30.4g)に水70mLを加えて溶かす。遮光して保存する。用時調製する。
ヨウ化カリウムデンプン紙 新たに調製したヨウ化カリウムデンプン試液にろ紙を浸し、清浄な室で乾燥して調製する。共栓瓶に入れ、光及び湿気を避けて保存する。
ヨウ化カリウムデンプン試液 ヨウ化カリウム0.5g(0.45~0.54g)を新たに調製したデンプン試液100mLに溶かす。用時調製する。
溶性デンプン 〔特級〕
溶性デンプン試液 溶性デンプン1g(0.5~1.4g)に冷水10mLを加え、よくすり混ぜ、熱湯90mL中に絶えずかき混ぜながら徐々に注ぎ込み、3分間緩やかに煮沸し、冷却する。用時調製する。
ヨウ素 I2〔特級〕
ヨウ素・アセトン試液 ヨウ素10g(9.5~10.4g)及びヨウ化カリウム6g(5.5~6.4g)を乳鉢で混和し、水10mLに溶かし、90%エタノールを加えて100mLとする。この溶液3.5mLを量り、アセトンを加えて100mLとする。
ヨウ素酸カリウム・デンプン紙 0.2%KIO3溶液及びデンプン試液の等容量混液に定量用ろ紙を浸し、暗所で風乾する。遮光し、密栓して保存する。
ヨウ素試液 ヨウ素14g(13.5~14.4g)をヨウ化カリウム溶液(2→5)100mLに溶かし、希塩酸1mL及び水を加えて1,000mLとする(0.05mol/L)。遮光して保存する。
ヨウ素酸カリウム KIO3〔特級〕
ヨウ素酸カリウム(標準試薬) KIO3〔容量分析用標準試薬〕
ヨウ素・ルゴール試液 ヨウ素5g(4.5~5.4g)及びヨウ化カリウム10g(9.5~10.4g)を乳鉢で混和し、水に溶かし、100mLとする。用時、水で5倍に希釈して用いる。
ライネッケ塩 NH4〔Cr(NH3)2(SCN)4〕H2O〔1級〕
ライネッケ塩試液 ライネッケ塩0.5g(0.45~0.54g)に水20mLを加え、1時間しばしば振り混ぜ、ろ過する。48時間以内に使用する。
酪酸 CH3CH2CH2COOH〔特級〕
n-酪酸ナトリウム CH3CH2CH2COONa 白色~ほとんど白色の粉末である。
溶状 本品の水溶液(1→20)は、透明である又は僅かに微濁する。
含量 90%以上
ラフィノース C18H32O16・5H2O
比旋光度 〔α〕D20=+122~+124°
重金属 10μg/g以下
水分 14~16%
ランタン・アリザリンコンプレクソン試液 アリザリンコンプレクソンとランタン塩に、ヘキサメチレンテトラミン・フタル酸水素カリウム系の緩衝剤を加えた混成試薬の10%水溶液とする。
リトマス紙、赤色 〔リトマス紙、赤色リトマス紙〕
リボフラビン C17H20N4O6〔日局〕
硫化アンモニウム試液 〔硫化アンモニウム溶液(無色)、1級〕 遮光した小瓶に全満して保存する。
硫化水素 H2S 無色の有毒ガスで空気より重く、水に溶ける。硫化鉄に希硫酸又は希塩酸を作用させて調製する。希酸を作用させるとき、硫化水素を発生するものであれば、他の硫化物を代用することができる。
硫化鉄 FeS〔1級〕
硫化ナトリウム Na2S・9H2O〔特級〕
硫化ナトリウム試液 次のいずれかの方法により調製する。
① 硫化ナトリウム5g(4.5~5.4g)を水10mL及びグリセリン30mLの混液に溶かす。
② 水酸化ナトリウム5g(4.5~5.4g)を水30mL及びグリセリン90mLの混液に溶かし、その半容量に冷時硫化水素を飽和し、それに残りの半容量を混和する。遮光した瓶にほとんど全満して保存する。調製した後、3か月以内に用いる。
硫酸 H2SO4〔特級〕 H2SO495.0%以上を含む。
硫酸、希 硫酸5.7mLを水10mLに注意しながら加え、放冷した後、水を加えて100mLとする(10%)。
硫酸亜鉛 (ZnSO4・7H2O)〔特級〕
硫酸アデニン (C5H5N5)2・H2SO4 白色の結晶である。水又はエタノールに溶けにくい。
溶状 本品0.2g(0.15~0.24g)に水20mLを加え、加温溶解するとき、溶液は、無色で、澄明である。
吸光度 本品0.1gを0.001gの桁まで量り、その数値を記録し、塩化カリウム・塩酸緩衝液に溶かし、10,000mLとし、この溶液につき、波長262nmで吸光度を測定するとき、E1cm1%=638~668である。
水分 6.7~11.1%(0.3g)
強熱残分 0.5%以下(10g)
硫酸アンモニウム (NH4)2SO4〔特級〕
硫酸試液、3mol/L 硫酸180mLを水1,000mL中にかき混ぜながら徐々に加えた後、放冷する。
硫酸試液、2.5mol/L 硫酸150mLを水1,000mL中にかき混ぜながら徐々に加えた後、放冷する。
硫酸試液、2mol/L 硫酸120mLを水1,000mL中にかき混ぜながら徐々に加えた後、放冷する。
硫酸試液、0.5mol/L 硫酸30mLを水1,000mL中にかき混ぜながら徐々に加えた後、放冷する。
硫酸試液、0.2mol/L 硫酸12mLを水1,000mL中にかき混ぜながら徐々に加えた後、放冷する。
硫酸試液、0.1mol/L 硫酸6mLを水1,000mL中にかき混ぜながら徐々に加えた後、放冷する。
硫酸、硫酸呈色物用 あらかじめ、次の方法で含量を測定した硫酸に注意して水を加えて硫酸(H2SO4)94.5~95.5%に調製する。保存中、水分を吸収して濃度が変わったときは、新たに調製する。
定量法 硫酸約2gを0.01gの桁まで量り、その数値を記録し、共栓フラスコ中に入れ、水30mLを加え、放冷した後、1mol/L水酸化ナトリウム溶液で滴定する(指示薬 ブロムチモールブルー試液2~3滴)。
1mol/L水酸化ナトリウム溶液1mL=49.04mgH2SO4
硫酸カリウム K2SO4〔特級〕
硫酸コバルト CoSO4・7H2O〔特級〕
硫酸水素カリウム KHSO4〔硫酸水素カリウム(酸性硫酸カリウム)、特級〕
硫酸第一鉄 FeSO4・7H2O〔特級〕
硫酸第一鉄試液 硫酸第一鉄8g(7.5~8.4g)に新たに煮沸し冷却した水100mLを加えて溶かす。用時調製する。
硫酸第一鉄アンモニウム FeSO4・(NH4)2SO4・6H2O〔硫酸第一鉄アンモニウム(モール塩)、特級〕
硫酸第二水銀試液 黄色酸化第二水銀5g(4.5~5.4g)に水40mLを加え、かき混ぜながら硫酸20mLを徐々に加え、更に水40mLを加え、溶けるまでかき混ぜる。
硫酸第二セリウムアンモニウム Ce(SO4)2・2(NH4)2SO4・4H2O〔特級〕
硫酸第二鉄 Fe2(SO4)3・nH2O〔特級〕
硫酸第二鉄アンモニウム Fe2(SO4)3・(NH4)2SO4・24H2O〔硫酸第二鉄アンモニウム(鉄ミョウバン)、特級〕
硫酸第二鉄アンモニウム試液 硫酸第二鉄アンモニウム8g(7.5~8.4g)に水を加えて溶かし、100mLとする。
硫酸呈色物用硫酸 硫酸、硫酸呈色物用の項に定める。
硫酸銅 CuSO4・5H2O〔特級〕
硫酸銅、無水 CuSO4〔硫酸銅(無水)、1級〕
硫酸銅・アンモニア試液 アンモニア試液及びクエン酸溶液(1→5)の混液(2:3)50mLに硫酸銅0.4g(0.35~0.44g)を溶かす。
硫酸銅試液 硫酸銅12.5g(12.45~12.54g)に水を加えて溶かし、100mLとする(0.5mol/L)。
硫酸ナトリウム、無水 Na2SO4〔硫酸ナトリウム(無水)、特級〕
硫酸ポリミキシンB 〔日局〕
硫酸マグネシウム MgSO4・7H2O〔特級〕
硫酸マグネシウム試液 硫酸マグネシウム12g(11.5~12.4g)に水を加えて溶かし、100mLとする(0.5mol/L)。
硫酸マンガン MnSO4・xH2O〔特級〕
硫酸リチウム Li2SO4・H2O〔特級〕
流動パラフィン パラフィン、流動の項に定める。
リリーの染色液 0.25mol/Lの塩化メチルロザニリン・エタノール試液20mL及びシュウ酸アンモニウム溶液(1→100)80mLを混和して溶かす。用時調製する。
リン酸 H3PO4〔特級〕
リン酸一水素ナトリウム・二水和物 Na2HPO4・2H2O
リン酸塩緩衝液、pH2.0 リン酸二水素ナトリウム31.2g(31.15~31.24g)に水を加えて溶かし、1,000mLとし、さらに、リン酸(1→50)を加えてpH2.0に調整する。
リン酸塩緩衝液、pH3.5 リン酸二水素ナトリウム7.8g(7.75~7.84g)に水を加えて溶かし、1,000mLとし、さらに、リン酸(3→500)を加えてpH3.5に調整する。
リン酸塩緩衝液、pH7.0 緩衝液用0.2mol/Lリン酸二水素カリウム試液50mL及び0.2mol/L水酸化ナトリウム試液29.54mLを混ぜ、水を加えて200mLとする。
リン酸塩緩衝液、pH7.0、比施光度測定用 1mol/Lリン酸二水素カリウム試液50mLに1mol/L水酸化ナトリウム試液を加えてpH7.0±0.1に調整し、水を加えて100mLとする。
リン酸塩緩衝液、pH7.5 リン酸二水素カリウム22.2g(22.15~22.24g)及びリン酸一水素カリウム177.8g(177.75~177.84g)に水を加えて溶かし、1,000mLとする。
リン酸塩緩衝液、pH8.0 リン酸二水素カリウム3.06g(3.055~3.064g)を水450mLに溶かし、1mol/L水酸化ナトリウム試液を加えてpHを8.0に調整した後、水を加えて500mLとする。
リン酸塩緩衝液、0.1mol/L、酵素力試験用 0.1mol/Lリン酸一水素ナトリウム試液に0.1mol/Lリン酸二水素カリウム試液を加えて所定のpHに調整する。
リン酸塩緩衝液、0.02mol/L、酵素力試験用 0.02mol/Lリン酸一水素ナトリウム試液に0.02mol/Lリン酸二水素カリウム試液を加えて所定のpHに調整する。
リン酸塩緩衝液・アセトニトリル試液 リン酸二水素カリウム2.0g(1.95~2.04g)に水600mL及びアセトニトリル400mLを加えて溶かし、1mol/L水酸化ナトリウム試液を加えてpH7に調整する。
リン酸塩・炭酸水素ナトリウム緩衝液 リン酸一水素カリウム16.73g(16.725~16.734g)、リン酸二水素カリウム0.523g(0.5225~0.5234g)及び炭酸水素ナトリウム20.0g(19.95~20.04g)に水を加えて溶かし、1,000mLとする。
リン酸・酢酸・ホウ酸・水酸化ナトリウム緩衝液 リン酸3.92g(3.915~3.924g)、酢酸2.4g(2.35~2.44g)及びホウ酸2.48g(2.475~2.484g)を水1,000mLに溶かしたもの40mLに0.2mol/L水酸化ナトリウム試液約28mLを加えてpH9.2に調整する。この溶液30mLにメタノール70mLを加え、よく混和する。
リン酸一水素カリウム K2HPO4〔リン酸二カリウム、特級〕
0.15mol/Lリン酸一水素カリウム試液 リン酸一水素カリウム26.13g(26.125~26.134g)に水を加えて溶かし、1,000mLとする。
リン酸一水素ナトリウム Na2HPO4・12H2O〔リン酸二ナトリウム(12水塩)、特級〕
リン酸一水素ナトリウム、無水 Na2HPO4〔リン酸二ナトリウム(無水)、特級〕
リン酸一水素ナトリウム、無水、pH測定用 Na2HPO4〔リン酸二ナトリウム(無水)、pH測定用〕
リン酸一水素ナトリウム試液 リン酸一水素ナトリウム12g(11.5~12.4g)に水を加えて溶かし、100mLとする(1mol/L)。
リン酸一水素ナトリウム試液、0.1mol/L 無水リン酸一水素ナトリウム14.2g(14.15~14.24g)に水を加えて溶かし、1,000mLとする。
リン酸一水素ナトリウム試液、0.05mol/L 無水リン酸一水素ナトリウム7.1g(7.05~7.14g)に水を加えて溶かし、1,000mLとする。
リン酸一水素ナトリウム試液、0.02mol/L 無水リン酸一水素ナトリウム2.84g(2.835~2.844g)に水を加えて溶かし、1,000mLとする。
リン酸二水素カリウム KH2PO4〔リン酸一カリウム、特級〕
リン酸二水素カリウム、pH測定用 KH2PO4〔リン酸一カリウム、pH測定用〕
リン酸二水素カリウム試液、1mol/L リン酸二水素カリウム136.1g(136.05~136.14g)に水を加えて溶かし、1,000mLとする。
リン酸二水素カリウム試液、0.1mol/L リン酸二水素カリウム13.61g(13.605~13.614g)に水を加えて溶かし、1,000mLとする。
リン酸二水素カリウム試液、0.02mol/L リン酸二水素カリウム2.72g(2.715~2.724g)に水を加えて溶かし、1,000mLとする。
0.2mol/Lリン酸二水素カリウム試液、緩衝液用 〔リン酸二水素カリウム、pH測定用〕 リン酸二水素カリウム27.22g(27.215~27.224g)に水を加えて溶かし、1,000mLとする。
リン酸二水素ナトリウム試液、pH4.5 リン酸二水素ナトリウム15.6g(15.55~15.64g)に水を加えて溶かし、1,000mLとし、必要ならば、1mol/L水酸化ナトリウム試液又はリン酸(1→10)を加えてpH4.5に調整する。
リンタングステン酸 P2O5・24WO3・nH2O〔特級〕
リンタングステン酸試液 リンタングステン酸1g(0.5~1.4g)に水を加えて溶かし、100mLとする。
リン発色試液 バナジン酸アンモニウム1.12g(1.115~1.124g)を量り、約300mLの水を加えて溶かし、硝酸250mLを加える。これにモリブデン酸アンモニウム27g(26.5~27.4g)を溶かした水溶液を加え、更に水を加えて1,000mLとする。
リンモリブデン酸 P2O5・24MoO3・nH2O〔特級〕
ルゴール液 ヨウ素1g(0.5~1.4g)及びヨウ化カリウム2g(1.5~2.4g)を乳鉢で混和し、これに水300mLを少量ずつかき混ぜながら加えて溶かす。
レソルシノール C6H4(OH)2〔特級〕
レソルシノール・硫酸試液 レソルシノール0.1gを硫酸(1→10)10mLに溶かす。
レゾルシン レソルシノールの項に定める。
(3) 容量分析用標準液
容量分析用標準液は、濃度が精密に知られた試薬溶液で、主として容量分析に用いるものである。
容量分析用標準液には、モル液を用いる。溶液1,000mL中に有効物質1グラム分子量を含む溶液を1モル液とし、1mol/Lで表す。また、必要に応じて、それらを一定の割合に薄めた溶液を用いる。
容量分析用標準液は、別に規定する場合を除き、無色又は遮光した共栓瓶に入れ保存する。
調製
容量分析用標準液は、次のいずれかの方法により調製し、モル濃度係数を定める。
① 純物質約1グラム分子量を有効数字3桁まで量り、その数値を記録し、溶媒を加えて溶かし、1,000mLとし、近似的濃度の1mol/L溶液を調製する。純物質が得られない場合は、純度が正確に判明している物質を用いることができる。
② 物質約1グラム分子量を量り、溶媒を加えて溶かし、約1,000mLとし、近似的濃度の1mol/L溶液を調製し、標定してモル濃度係数を定めた後、使用する。
標定は、モル濃度係数を定める操作であり、標準物質を有効数字3桁まで量り、その数値を記録し、溶媒を加えて溶かし、未標定モル液で滴定し、そのモル濃度係数fを求める。
f=1000a/(V×E×c)
E:標準物質の分子量(g)
a:標準物質の採取量(g)
V:未標定モル液の消費量(mL)
c:モル濃度
直接に標準物質を用いない場合は、モル濃度係数既知のモル液を用いて未標定モル液を標定する。
f2=(V1×f1)/V2
f1:モル濃度係数既知のモル液のモル濃度係数
f2:未標定モル液のモル濃度係数
V1:モル濃度係数既知のモル液の量(mL)
V2:未標定モル液の量(mL)
③ モル濃度係数既知のモル液の一定容量を正確に薄め、調製する。
0.1mol/L亜硝酸ナトリウム液
1,000mL中亜硝酸ナトリウム(NaNO2:69.00)6.900gを含む。
調製 亜硝酸ナトリウム7.2g(7.15~7.24g)に水を加えて溶かし、1,000mLとし、次の標定を行う。
標定 スルファミン酸(標準試薬)をデシケーター(減圧,シリカゲル)で48時間乾燥し、その約0.25gを0.001gの桁まで量り、その数値を記録し、塩酸5mL及び水50mLを加えて溶かし、15℃以下に冷却した後、砕氷25g(24.5~25.4g)を加え、かき混ぜながら調製した亜硝酸ナトリウム溶液で滴定し、モル濃度係数を計算する。この場合において、滴定の終点は、亜硝酸ナトリウム溶液を滴加して1分後に、被滴定液をガラス棒に付け、その先端でヨウ化亜鉛デンプン紙に触れるとき、30秒以内に青色を呈するときとする。
0.1mol/L亜硝酸ナトリウム溶液1mL=9.709mgHOSO2NH2
注意:遮光して保存する。長く保存したものは、標定し直して用いる。
0.05mol/Lエチレンジアミン四酢酸二ナトリウム溶液
1,000mL中エチレンジアミン四酢酸二ナトリウム(C10H14N2Na2O8・2H2O:372.24)18.61gを含む。
調製 エチレンジアミン四酢酸二ナトリウム19g(18.5~19.4g)に水を加えて溶かし、1,000mLとし、次の標定を行う。
標定 亜鉛(標準試薬)を希塩酸で洗い、次に、水洗し、さらに、アセトンで洗った後、110℃で5分間乾燥する。その後、デシケーター(シリカゲル)で放冷し、その約0.8gを0.001gの桁まで量り、その数値を記録し、希塩酸12mL及び臭素試液5滴を加え、穏やかに加温して溶かす。煮沸して過量の臭素を追い出した後、200mLの全量フラスコに入れ、水を標線まで加えて200mLとする。この溶液20mLを全量ピペットを用いて量り、水酸化ナトリウム溶液(1→50)を加えて中性とし、pH10.7のアンモニア・塩化アンモニウム緩衝液5mL及びエリオクロムブラックT・塩化ナトリウム指示薬0.04g(0.035~0.044g)を加え、調製したエチレンジアミン四酢酸二ナトリウム溶液で、溶液の赤紫色が青紫色に変わるまで滴定し、モル濃度係数を計算する。
0.05mol/Lエチレンジアミン四酢酸二ナトリウム溶液1mL=3.269mgZn
注意:ポリエチレン瓶に保存する。
0.02mol/Lエチレンジアミン四酢酸二ナトリウム溶液
1,000mL中エチレンジアミン四酢酸二ナトリウム(C10H14N2Na2O8・2H2O:372.24)7.445gを含む。
調製 エチレンジアミン四酢酸二ナトリウム7.5g(7.45~7.54g)に水を加えて溶かし、1,000mLとし、次の標定を行う。
標定 亜鉛(標準試薬)を希塩酸で洗い、次に、水洗し、さらに、アセトンで洗った後、110℃で5分間乾燥する。その後、デシケーター(シリカゲル)で放冷し、その約0.3gを0.001gの桁まで量り、その数値を記録し、希塩酸5mL及び臭素試液5滴を加え、穏やかに加温して溶かす。煮沸して過量の臭素を追い出した後、200mLの全量フラスコに入れ、水を標線まで加えて200mLとする。この溶液20mLを全量ピペットを用いて量り、水酸化ナトリウム溶液(1→50)を加えて中性とする。pH10.7のアンモニア・塩化アンモニウム緩衝液5mL及びエリオクロムブラックT・塩化ナトリウム指示薬0.04g(0.035~0.044g)を加え、調製したエチレンジアミン四酢酸二ナトリウム溶液で、溶液の赤紫色が青紫色に変わるまで滴定し、モル濃度係数を計算する。
0.02mol/Lエチレンジアミン四酢酸二ナトリウム液1mL=1.308mgZn
注意:ポリエチレン瓶に保存する。
0.01mol/Lエチレンジアミン四酢酸二ナトリウム溶液
1,000mL中エチレンジアミン四酢酸二ナトリウム(C10H14N2Na2O8・2H2O:372.24)3.722gを含む。
調製 用時、0.02mol/Lエチレンジアミン四酢酸二ナトリウム溶液に水を加えて正確に2倍容量とする。
0.05mol/L塩化マグネシウム溶液
1,000mL中塩化マグネシウム(MgCl2・6H2O:203.30)10.17gを含む。
調製 塩化マグネシウム10.2g(10.15~10.24g)に新たに煮沸し冷却した水を加えて溶かし、1,000mLとし、次の標定を行う。
標定 調製した塩化マグネシウム溶液25mLを全量ピペットを用いて量り、水50mL、pH10.7のアンモニア・塩化アンモニウム緩衝液3mL及びエリオクロムブラックT・塩化ナトリウム指示薬0.04g(0.035~0.044g)を加え、0.05mol/Lエチレンジアミン四酢酸二ナトリウム溶液で滴定し、モル濃度係数を計算する。この場合において、滴定の終点は、溶液の赤紫色が青紫色に変わるときとする。なお、終点近くでは、ゆっくり滴定する。
1mol/L塩酸
1,000mL中塩酸(HCl:36.46)36.46gを含む。
調製 塩酸90mLに水を加えて1,000mLとし、次の標定を行う。
標定 炭酸ナトリウム(標準試薬)を500~650℃で40~50分間加熱した後、デシケーター(シリカゲル)で放冷し、その約1.3gを0.01gの桁まで量り、水50mLを加えて溶かし、メチルレッド試液3滴を加え、調製した塩酸で滴定し、モル濃度係数を計算する。この場合において、滴定の終点は、溶液を注意して煮沸し、緩く栓をして冷却するとき、持続して橙色~橙赤色を呈するときとする。
1mol/L塩酸1mL=53.00mgNa2CO3
0.1mol/L塩酸
1,000mL中塩酸(HCl:36.46)3.646gを含む。
調製 塩酸9.0mLに水を加えて1,000mLとし、次の標定を行う。
標定 炭酸ナトリウム(標準試薬)を500~650℃で40~50分間加熱した後、デシケーター(シリカゲル)で放冷し、その約0.15gを0.001gの桁まで量り、水30mLを加えて溶かし、メチルレッド試液3滴を加え、調製した塩酸で滴定し、モル濃度係数を計算する。この場合において、滴定の終点は、溶液を注意して煮沸し、緩く栓をして冷却するとき、持続して橙色~橙赤色を呈するときとする。
0.1mol/L塩酸1mL=5.300mgNa2CO3
0.05mol/L塩酸
1,000mL中塩酸(HCl:36.46)1.823gを含む。
調製 用時、0.1mol/L塩酸に水を加えて正確に2倍容量とする。
0.01mol/L塩酸
1,000mL中塩酸(HCl:36.46)0.3646gを含む。
調製 用時、0.1mol/L塩酸に水を加えて正確に10倍容量とする。
0.001mol/L塩酸
1,000mL中塩酸(HCl:36.46)0.03646gを含む。
調製 用時、0.1mol/L塩酸に水を加えて正確に100倍容量とする。
0.1mol/L過塩素酸
1,000mL中過塩素酸(HClO4:100.46)10.05gを含む。
調製 過塩素酸8.5mLを非水滴定用氷酢酸800mL中に30℃以下に保ちながら徐々に加える。数時間放置した後、無水酢酸22.2mLを加え、振り混ぜ、非水滴定用氷酢酸を追加して1,000mLとし、48時間放置し、次の標定を行う。
標定 フタル酸水素カリウム(標準試薬)を105℃で4時間乾燥した後、デシケーター(シリカゲル)で放冷し、その約0.5gを0.001gの桁まで量り、その数値を記録し、非水滴定用氷酢酸80mLを加えて溶かし、塩化メチルロザニリン試液3滴を加え、調製した過塩素酸で青色を呈するまで滴定し、モル濃度係数を計算する。同様の方法で空試験を行い補正する。
0.1mol/L過塩素酸1mL=20.42mgKHC6H4(COO)2
注意:湿気を避けて保存する。
0.1mol/L過塩素酸・ジオキサン溶液
1,000mL中過塩素酸(HClO4:100.46)10.05gを含む。
調製 過塩素酸8.5mLにジオキサンを加えて1,000mLとし、次の標定を行う。
標定 フタル酸水素カリウム(標準試薬)を105℃で4時間乾燥した後、デシケーター(シリカゲル)で放冷し、その約0.5gを0.001gの桁まで量り、非水滴定用氷酢酸80mLを加えて溶かし、塩化メチルロザニリン試液3滴を加え、調製した過塩素酸・ジオキサン溶液で青色を呈するまで滴定し、モル濃度係数を計算する。同様の方法で空試験を行い補正する。
0.1mol/L過塩素酸・ジオキサン溶液1mL=20.42mgKHC6H4(COO)2
注意:湿気を避け、冷所に保存する。
0.02mol/L過マンガン酸カリウム溶液
1,000mL中過マンガン酸カリウム(KMnO4:158.03)3.161gを含む。
調製 過マンガン酸カリウム3.2g(3.15~3.24g)に水を加えて溶かし、1,000mLとし、15分間煮沸して密栓し、48時間以上放置した後、ガラスろ過器(G3又はG4)を用いてろ過し、次の標定を行う。
標定 シュウ酸ナトリウム(標準試薬)を150~200℃で1~1.5時間乾燥した後、デシケーター(シリカゲル)で放冷し、その約0.3gを0.001gの桁まで量り、その数値を記録し、500mLの三角フラスコに入れ、水30mLを加えて溶かす。次に、硫酸(1→20)250mLを加え、液温を30~35℃とし、調製した過マンガン酸カリウム溶液をビュレットに入れ、緩やかにかき混ぜながら、その40mLを30秒以内に加え、溶液の紅色が消えるまで放置する。その後、55~60℃に加温して滴定を続け、30秒間持続して淡紅色を呈するまで滴定し、モル濃度係数を計算する。この場合において、終点前の0.5~1mLは注意して滴加し、過マンガン酸カリウム液の色が消えてから次の1滴を加える。
0.02mol/L過マンガン酸カリウム溶液1mL=6.700mgNa2C2O4
注意:遮光して保存する。長く保存したものは、標定し直して用いる。
0.05mol/L酢酸亜鉛溶液
1,000mL中酢酸亜鉛〔Zn(CH3COO)2・2H2O:219.50〕10.98gを含む。
調製 酢酸亜鉛11.1g(11.05~11.14g)に水40mL及び希酢酸4mLを加えて溶かし、水を加えて1,000mLとし、次の標定を行う。
標定 0.05mol/Lエチレンジアミン四酢酸二ナトリウム溶液20mLを全量ピペットを用いて量り、水50mL、pH10.7のアンモニア・塩化アンモニウム緩衝液3mL及びエリオクロムブラックT・塩化ナトリウム指示薬0.04g(0.035~0.044g)を加え、調製した酢酸亜鉛溶液で滴定し、モル濃度係数を計算する。滴定の終点は、溶液の青色が青紫色に変わるときとする。
0.005mol/L酢酸第二水銀溶液
1,000mL中酢酸第二水銀〔Hg(CH3COO)2:318.68〕1.593gを含む。
調製 酢酸第二水銀1.6g(1.55~1.64g)に希硝酸(1→10)60mLを加えて溶かし、水を加えて1,000mLとし、次の標定を行う。
標定 塩化ナトリウム(標準試薬)を500~650℃で40~50分間乾燥した後、デシケーター(シリカゲル)で放冷し、その約0.58gを0.001gの桁まで量り、その数値を記録し、水を加えて溶かし、1,000mLの全量フラスコに入れ、更に水を標線まで加えて1,000mLとする。この溶液20mLを全量ピペットを用いて量り、ブロムフェノールブルー試液1滴を加え、溶液が黄色を呈するまで希硝酸を滴加した後、希硝酸5mL、メタノール100mL及びジフェニルカルバゾン試液1mLを加え、よく振り混ぜながら、調製した酢酸第二水銀溶液で、溶液の淡黄色が赤紫色に変わるまで滴定し、モル濃度係数を計算する。
0.005mol/L酢酸第二水銀溶液1mL=0.5844mgNaCl
0.017mol/L重クロム酸カリウム溶液
1,000mL中重クロム酸カリウム(K2Cr2O7:294.18)4.903gを含む。
調製 重クロム酸カリウム(標準試薬)を粉末とし、100~110℃で3~4時間乾燥した後、デシケーター(シリカゲル)で放冷し、その約4.903gを0.001gの桁まで量り、その数値を記録し、水を加えて溶かし、1,000mLの全量フラスコに入れ、更に水を標線まで加えて1,000mLとし、モル濃度係数を計算する。
0.05mol/L臭素溶液
1,000mL中臭素(Br2:159.80)7.990gを含む。
調製 臭素酸カリウム2.8g(2.75~2.84g)及び臭化カリウム15g(14.5~15.4g)に水を加えて溶かし、1,000mLとし、次の標定を行う。
標定 調製した臭素液25mLをヨウ素瓶中に全量ピペットを用いて量り、水120mL、次に、塩酸5mLを30秒以内に加える。塩酸を加えてから30秒以内に密栓し、穏やかに振り混ぜ、5分間放置した後、遊離したヨウ素を0.1mol/Lチオ硫酸ナトリウム溶液で滴定し、モル濃度係数を計算する。この場合において、滴定の終点は、溶液が終点近くで黄色になったとき、デンプン試液3mLを加え、生じた青色が脱色するときとする。同様の方法で空試験を行い補正する。
0.1mol/L硝酸銀溶液
1,000mL中硝酸銀(AgNO3:169.87)16.99gを含む。
調製 硝酸銀17.0g(16.95~17.04g)に水を加えて溶かし、1,000mLとし、次の標定を行う。
標定 塩化ナトリウム(標準試薬)を500~650℃で40~50分間乾燥した後、デシケーター(シリカゲル)で放冷し、その約0.15gを0.001gの桁まで量り、その数値を記録し、水50mLを加えて溶かし、クロム酸カリウム試液1mLを加え、振り動かしながら、調製した硝酸銀溶液で持続して淡赤褐色を呈するまで滴定し、モル濃度係数を計算する。
0.1mol/L硝酸銀溶液1mL=5.844mgNaCl
注意:遮光して保存する。
0.1mol/L水酸化カリウム溶液
1,000mL中水酸化カリウム(KOH:56.11)5.611gを含む。
調製 水酸化カリウム6.5g(6.45~6.54g)に水950mLを加えて溶かし、これに、新たに調製した飽和水酸化バリウム溶液を沈殿が生じなくなるまで滴加し、溶液をよく混ぜて、密栓し、24時間放置した後、上澄液を傾斜し、又はガラスろ過器(G3又はG4)を用いてろ過し、次の標定を行う。
標定 スルファミン酸(標準試薬)をデシケーター(減圧,シリカゲル)で約48時間乾燥し、その約0.25gを0.001gの桁まで量り、その数値を記録し、新たに煮沸し冷却した水25mLを加えて溶かし、ブロムチモールブルー試液2滴を加え、調製した水酸化カリウム溶液で緑色を呈するまで滴定し、モル濃度係数を計算する。
0.1mol/L水酸化カリウム溶液1mL=9.709mgHOSO2NH2
注意:密栓した瓶又は二酸化炭素吸収管(ソーダ石灰管)を付けた瓶に保存する。長く保存したものは、標定し直して用いる。
0.5mol/L水酸化カリウム・エタノール溶液
1,000mL中水酸化カリウム(KOH:56.11)28.06gを含む。
調製 水酸化カリウム35g(34.5~35.4g)に水20mLを加えて溶かし、無アルデヒドエタノールを加えて1,000mLとし、密栓し、24時間放置した後、上澄液を30秒以内に傾斜してとり、次の標定を行う。
標定 0.25mol/L硫酸25mLを全量ピペットを用いて量り、水50mL及びフェノールフタレイン試液2滴を加え、調製した水酸化カリウム・エタノール溶液で淡赤色を呈するまで滴定し、モル濃度係数を計算する。
注意:遮光した瓶に密栓して保存する。標定は、用時行う。
0.1mol/L水酸化カリウム・エタノール溶液
1,000mL中水酸化カリウム(KOH:56.11)5.611gを含む。
調製 水酸化カリウム7g(6.5~7.4g)に、水20mLを加えて溶かし、無アルデヒドエタノールを加えて1,000mLとし、密栓し、24時間放置した後、上澄液を30秒以内に傾斜してとり、次の標定を行う。
標定 0.05mol/L硝酸25mLを全量ピペットを用いて量り、水50mL及びフェノールフタレイン試液2滴を加え、調製した水酸化カリウム・エタノール溶液で淡赤色を呈するまで滴定し、モル濃度係数を計算する。
注意:遮光した瓶に密栓して保存する。標定は、用時行う。
1mol/L水酸化ナトリウム溶液
1,000mL中水酸化ナトリウム(NaOH:40.00)40.00gを含む。
調製 水酸化ナトリウム42g(41.5~42.4g)に水950mLを加えて溶かし、これに新たに調製した水酸化バリウム飽和溶液を沈殿が生じなくなるまで滴加し、溶液をよく混ぜ、密栓し、24時間放置した後、上澄液を傾斜し、又はガラスろ過器(G3又はG4)を用いてろ過し、次の標定を行う。
標定 スルファミン酸(標準試薬)をデシケーター(減圧,シリカゲル)で約48時間乾燥し、その約2.5gを0.01gの桁まで量り、その数値を記録し、新たに煮沸し冷却した水25mLを加えて溶かし、ブロムチモールブルー試液2滴を加え、調製した水酸化ナトリウム溶液で緑色を呈するまで滴定し、モル濃度係数を計算する。
1mol/L水酸化ナトリウム溶液1mL=97.09mgHOSO2NH2
注意:密栓した瓶又は二酸化炭素吸収管(ソーダ石灰)を付けた瓶に保存する。長く保存したものは、標定し直して用いる。
0.1mol/L水酸化ナトリウム溶液
1,000mL中水酸化ナトリウム(NaOH:40.00)4.000gを含む。
調製 水酸化ナトリウム4.5g(4.45~4.54g)を量り、1mol/L水酸化ナトリウム溶液に準じて調製し、次の標定を行う。
標定 スルファミン酸(標準試薬)をデシケーター(減圧,シリカゲル)で約48時間乾燥し、その約0.25gを0.001gの桁まで量り、その数値を記録し、新たに煮沸し冷却した水25mLを加えて溶かし、ブロムチモールブルー試液2滴を加え、調製した水酸化ナトリウム溶液で緑色を呈するまで滴定し、モル濃度係数を計算する。
0.1mol/L水酸化ナトリウム溶液1mL=9.709mgHOSO2NH2
注意:1mol/L水酸化ナトリウム溶液に準じて保存する。長く保存したものは、標定し直して用いる。
0.1mol/Lチオシアン酸アンモニウム溶液
1,000mL中チオシアン酸アンモニウム(NH4SCN:76.12)7.612gを含む。
調製 チオシアン酸アンモニウム8g(7.5~8.4g)に水を加えて溶かし、1,000mLとし、次の標定を行う。
標定 0.1mol/L硝酸銀溶液25mLを全量ピペットを用いて量り、水50mL、硝酸2mL及び硝酸第二鉄アンモニウム試液2mLを加え、振り動かしながら、調製したチオシアン酸アンモニウム溶液で持続して赤褐色を呈するまで滴定し、モル濃度係数を計算する。
注意:遮光して保存する。
0.1mol/Lチオ硫酸ナトリウム溶液
1,000mL中チオ硫酸ナトリウム(Na2S2O3・5H2O:248.18)24.82gを含む。
調製 チオ硫酸ナトリウム25g(24.5~25.4g)及び無水炭酸ナトリウム0.2g(0.15~0.24g)に、新たに煮沸し冷却した水を加えて溶かし、1,000mLとし、24時間放置した後、次の標定を行う。
標定 ヨウ素酸カリウム(標準試薬)を120~140℃で1.5~2時間乾燥した後、デシケーター(シリカゲル)で放冷し、その約0.1gを0.0001gの桁までヨウ素瓶に量り、その数値を記録し、水25mLを加えて溶かし、ヨウ化カリウム2g(1.5~2.4g)及び希硫酸10mLを加え、密栓し、10分間放置した後、水100mLを加え、遊離したヨウ素を調製したチオ硫酸ナトリウム溶液で滴定し、モル濃度係数を計算する。この場合において、滴定の終点は、溶液が終点近くで淡黄色になったとき、デンプン試液3mLを加え、生じた青色が脱色するときとする。同様の方法で空試験を行い補正する。
0.1mol/Lチオ硫酸ナトリウム溶液1mL=3.567mgKIO3
注意:長く保存したものは、標定し直して用いる。
0.05mol/Lチオ硫酸ナトリウム溶液
1,000mL中チオ硫酸ナトリウム(Na2S2O3・5H2O:248.18)12.41gを含む。
調製 用時、0.1mol/Lチオ硫酸ナトリウム溶液に、新たに煮沸し冷却した水を加えて正確に2倍容量とする
0.01mol/Lチオ硫酸ナトリウム溶液
1,000mL中チオ硫酸ナトリウム(Na2S2O3・5H2O:248.18)2.482gを含む。
調製 用時、0.1mol/Lチオ硫酸ナトリウム溶液に、新たに煮沸し冷却した水を加えて正確に10倍容量とする。
0.005mol/Lチオ硫酸ナトリウム溶液
1,000mL中チオ硫酸ナトリウム(Na2S2O3・5H2O:248.18)1.241gを含む。
調製 用時、0.1mol/Lチオ硫酸ナトリウム溶液に、新たに煮沸し冷却した水を加えて正確に20倍容量とする。
0.1mol/Lナトリウムメトキシド溶液
1,000mL中ナトリウムメトキシド(CH3ONa:54.02)5.402gを含む。
調製 金属ナトリウムの新しい切片2.5g(2.45~2.54g)を氷冷したメタノール150mL中に少量ずつ加えて溶かした後、ベンゼンを加えて1,000mLとし、次の標定を行う。
標定 安息香酸をデシケーター(シリカゲル)で24時間乾燥し、その約0.3gを0.001gの桁まで量り、その数値を記録し、ジメチルホルムアミド80mLを加えて溶かし、チモールブルー・ジメチルホルムアミド試液3滴を加え、調製したナトリウムメトキシド溶液で青色を呈するまで滴定し、モル濃度係数を計算する。同様の方法で空試験を行い補正する。
0.1mol/Lナトリウムメトキシド溶液1mL=12.21mgC6H5COOH
注意:湿気を避けて、冷所に保存する。標定は、用時行う。
0.1mol/Lマグネシウムエチレンジアミン四酢酸二ナトリウム溶液
塩化マグネシウム(MgCl2・6H2O)20.33g(20.325~20.334g)に新たに煮沸し冷却した水300mLを加えて溶かし、別に、エチレンジアミン四酢酸二ナトリウム(C10H14N2Na2O8・2H2O)37.22g(37.215~37.224g)に水300mLを加えて溶かした溶液を混和し、1mol/L水酸化ナトリウム試液でpH7.0~8.0に合わせ、水を加えて正確に1,000mLとする。
0.05mol/Lヨウ素溶液
1,000mL中ヨウ素(I2:253.80)12.69gを含む。
調製 ヨウ素13g(12.5~13.4g)にヨウ化カリウム溶液(2→5)100mLを加えて溶かし、希塩酸1mL及び水を加えて1,000mLとし、次の標定を行う。
標定 次のいずれかの方法によりモル濃度係数を計算する。
① 三酸化ヒ素(標準試薬)を粉末とし、105℃で3~4時間乾燥した後、デシケーター(シリカゲル)で放冷し、その約0.15gを0.001gの桁まで量り、その数値を記録し、水酸化ナトリウム溶液(1→25)20mLを加え、必要ならば、加温して溶かす。水40mL及びメチルオレンジ試液2滴を加え、溶液が淡赤色になるまで希塩酸を加えた後、炭酸水素ナトリウム2g(1.5~2.4g)、水50mL及びデンプン試液3mLを加え、調製したヨウ素溶液を徐々に滴加し、溶液が持続して青色を呈するまで滴定し、モル濃度係数を計算する。
0.05mol/Lヨウ素溶液1mL=4.946mgAs2O3
② 調製したヨウ素溶液15mLを正確に量り、0.1mol/Lチオ硫酸ナトリウム溶液で滴定し、モル濃度係数を計算する(指示薬 デンプン試液又は電位差滴定法)。指示薬を用いる場合において、滴定の終点は、溶液が終点近くで淡黄色になったとき、デンプン試液3mLを加え、生じた青色が脱色するときとする。電位差滴定法の場合において、電極としては白金電極を用いる。
注意:遮光して保存する。長く保存したものは、標定し直して用いる。
0.05mol/Lヨウ素酸カリウム溶液
1,000mL中ヨウ素酸カリウム(KIO3:214.00)10.70gを含む。
調製 ヨウ素酸カリウム(標準試薬)を120~140℃で1.5~2時間乾燥した後、デシケーター(シリカゲル)で放冷し、その約10.70gを0.001gの桁まで量り、その数値を記録し、1,000mLの全量フラスコに入れ、水を加えて溶かし、更に水を標線まで加えて1,000mLとし、モル濃度係数を計算する。
0.5mol/L硫酸
1,000mL中硫酸(H2SO4:98.08)49.04gを含む。
調製 硫酸30mLを水1,000mL中にかき混ぜながら徐々に加え、放冷し、次の標定を行う。
標定 炭酸ナトリウム(標準試薬)を500~650℃で40~50分間加熱した後、デシケーター(シリカゲル)で放冷し、その約1.3gを0.01gの桁まで量り、その数値を記録し、水50mLを加えて溶かし、メチルレッド試液3滴を加え、調製した硫酸で滴定し、モル濃度係数を計算する。この場合において、滴定の終点は、溶液を注意して煮沸し、緩く栓をして冷却するとき、持続して橙色~橙赤色を呈するときとする。
0.5mol/L硫酸1mL=53.00mgNa2CO3
0.05mol/L硫酸
1,000mL中硫酸(H2SO4:98.08)4.904gを含む。
調製 硫酸3mLを水1,000mL中にかき混ぜながら徐々に加え、放冷し、次の標定を行う。
標定 炭酸ナトリウム(標準試薬)を500~650℃で40~50分間加熱した後、デシケーター(シリカゲル)で放冷し、その約0.15gを0.001gの桁まで量り、その数値を記録し、水30mLを加えて溶かし、メチルレッド試液3滴を加え、調製した硫酸で滴定し、モル濃度係数を計算する。この場合において、滴定の終点は、溶液を注意して煮沸し、緩く栓をして冷却するとき、持続して橙色~橙赤色を呈するときとする。
0.05mol/L硫酸1mL=5.300mgNa2CO3
0.01mol/L硫酸
1,000mL中硫酸(H2SO4:98.08)0.9808gを含む。
調製 用時、0.05mol/L硫酸に水を加えて正確に5倍容量とする。
0.005mol/L硫酸
1,000mL中硫酸(H2SO4:98.08)0.4904gを含む。
調製 用時、0.05mol/L硫酸に水を加えて正確に10倍容量とする。
0.1mol/L硫酸第二セリウムアンモニウム液
1,000mL中硫酸第二セリウムアンモニウム〔Ce(SO4)2・2(NH4)2SO4・4H2O:668.56〕66.86gを含む。
調製 硫酸第二セリウムアンモニウム68g(67.5~68.4g)に0.5mol/L硫酸を加えて溶かし、1,000mLとし、24時間放置した後、必要ならば、ガラスろ過器(G3又はG4)を用いてろ過し、次の標定を行う。
標定 調製した硫酸第二セリウムアンモニウム液25mLをヨウ素瓶に全量ピペットを用いて量り、水20mL及び希硫酸20mLを加え、次に、ヨウ化カリウム1g(0.5~1.4g)を加えて溶かし、30秒以内に0.1mol/Lチオ硫酸ナトリウム溶液で滴定し、モル濃度係数を計算する。この場合において、滴定の終点は、溶液が終点近くで淡黄色になったとき、デンプン試液3mLを加え、生じた青色が脱色するときとする。同様の方法で空試験を行い補正する。
注意:遮光して保存する。長く保存したものは、標定し直して用いる。
0.01mol/L硫酸第二セリウムアンモニウム溶液
1,000mL中硫酸第二セリウムアンモニウム〔Ce(SO4)2・2(NH4)2SO4・4H2O:668.56〕6.686gを含む。
調製 用時、0.1mol/L硫酸第二セリウムアンモニウム溶液に0.5mol/L硫酸を加えて正確に10倍容量とする。
(4) 標準液
標準液は、飼料添加物の試験において、試験の比較の基礎として用いる液である。
亜鉛標準液 硫酸亜鉛(ZnSO4・7H2O)4.40g(4.395~4.404g)に水を加えて溶かし、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとし、この溶液10mLを全量ピペットを用いて量り、1,000mLの全量フラスコに入れ、これに水を標線まで加えて1,000mLとする。この溶液1mLは、亜鉛(Zn)0.01mgを含む。
アルミニウム標準液 アルミニウム1.0g(0.95~1.04g)を量り、1,000mLの全量フラスコに入れ、塩酸(1→2)60mLを加え、加熱して溶かす。放冷後、水を標線まで加えて1,000mLとする。
アンモニウム標準液 塩化アンモニウム2.97g(2.965~2.974g)に水を加えて溶かし、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。この溶液10mLを全量ピペットを用いて量り、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。この溶液1mLは、アンモニウム(NH4)0.01mgを含む。
塩酸ジメチルアミン標準液 塩酸ジメチルアミン1.116g(1.1155~1.1164g)を水に溶かし、1,000mLとした後、その1mLをとり、水を加えて1,000mLとする。この溶液1mLは、ジメチルホルムアミド1μgに対応する。
カリウム標準液 塩化カリウム〔特級〕を400~500℃で40~50分間乾燥した後、デシケーター(シリカゲル)で放冷し、その1.907g(1.9065~1.9074g)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。この溶液1mLは、カリウム(K)1mgを含む。
カルシウム標準液 炭酸カルシウム〔特級〕を180℃で4時間乾燥し、その0.2500g(0.24995~0.25004g)を量り、希塩酸2mL及び水60mLを加えて溶かし、アンモニア試液でpHを6.0~7.0に調整し、100mLのメスフラスコに入れ、水を標線まで加えて100mLとする。
原子吸光光度用鉛標準液 鉛標準液、原子吸光光度用の項に定める。
ジチゾン用鉛標準液 一般試験法の鉛試験法に定めるところによるものとする。
シュウ酸塩pH標準液 pH標準液、シュウ酸塩の項に定める。
水酸化カルシウムpH標準液 pH標準液、水酸化カルシウムの項に定める。
炭酸塩pH標準液 pH標準液、炭酸塩の項に定める。
トリメチルアミン標準液 トリメチルアミン塩酸塩〔特級〕を105℃で4時間乾燥し、その80.84mgを0.001mgの桁まで量り、その数値を記録し、水を加えて溶かし、100mLの全量フラスコに入れ、更に水を標線まで加えて100mLとする。その10mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、水を標線まで加えて100mLとし、さらに、その10mLを全量ピペットを用いて量り、50mLの全量フラスコに入れ、水を標線まで加えて50mLとする。この溶液1mLは、トリメチルアミン10.0μgを含む。
鉄標準液 硫酸第二鉄アンモニウム86.34mg(86.335~86.344g)を量り、水100mLを加えて溶かし、1,000mLの全量フラスコに入れ、希塩酸5mL及び水を標線まで加えて1,000mLとする。この溶液1mLは、鉄(Fe)0.01mgを含む。
銅標準原液 硫酸銅(CuSO4・5H2O)3.929g(3.9285~3.9294g)を量り、希硝酸(3→5)を加えて溶かし、1,000mLの全量フラスコに入れ、希硝酸(3→5)を標線まで加えて1,000mLとする。
銅標準液 銅標準原液5mLを全量ピペットを用いて量り、200mLの全量フラスコに入れ、希硝酸(1→3)を標線まで加えて200mLとする。用時調製する。この溶液1mLは、銅(Cu)0.025mgを含む。
ナトリウム標準液 塩化ナトリウム(標準試薬)を500~650℃で40~50分間乾燥した後、デシケーター(シリカゲル)で放冷し、その2.542g(2.5415~2.5424g)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。この溶液1mLは、ナトリウム(Na)1mgを含む。
鉛標準原液 硝酸鉛159.9mg(159.85~159.94mg)を量り、希硝酸10mLを加えて溶かし、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。この溶液の調製及び保存には、可溶性鉛塩を含まないガラス容器を用いる。
鉛標準液 鉛標準原液10mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、水を標線まで加えて100mLとする。用時調製する。この溶液1mLは、鉛(Pb)0.01mgを含む。
鉛標準液、原子吸光光度用 鉛標準原液25mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、希硝酸(1→3)を標線まで加えて100mLとする。用時調製する。この溶液1mLは、鉛(Pb)0.025mgを含む。
鉛標準液、ジチゾン用 一般試験法の鉛試験法に定めるところによるものとする。
pH標準液、シュウ酸塩 pH測定用四シュウ酸カリウムを粉末とし、デシケーター(シリカゲル)で乾燥した後、その12.71g(12.705~12.714g)(0.05グラム分子量)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。
pH標準液、水酸化カルシウム pH測定用水酸化カルシウムを粉末とし、その5g(4.5~5.4g)をフラスコに入れ、水1,000mLを加え、よく振り混ぜ、23~27℃とし、十分に飽和した後、その温度で上澄液をろ過し、澄明なろ液(約0.02mol/L)を用いる。
pH標準液、炭酸塩 pH測定用炭酸水素ナトリウムをデシケーター(シリカゲル)で恒量になるまで乾燥し、その2.100g(0.025グラム分子量)及びpH測定用炭酸ナトリウムを300~500℃で恒量になるまで乾燥し、その2.650g(2.6495~2.6504g)(0.025グラム分子量)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。
pH標準液、フタル酸塩 pH測定用フタル酸水素カリウムを粉末にし、110℃で恒量になるまで乾燥し、その10.21g(10.205~10.214g)(0.05グラム分子量)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。
pH標準液、ホウ酸塩 pH測定用ホウ酸ナトリウムをデシケーター(水で潤した臭化ナトリウム)中に放置し、恒量とした後、その3.814g(3.8135~3.8144g)(0.01グラム分子量)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、更に水を標線まで加えて1,000mLとする。
pH標準液、リン酸塩 pH測定用リン酸二水素カリウム及びpH測定用無水リン酸一水素ナトリウムを粉末にし、110℃で恒量になるまで乾燥し、リン酸二水素カリウム3.402g(3.4015~3.4024g)(0.025グラム分子量)及びリン酸一水素ナトリウム3.549g(3.5485~3.5494g)(0.025グラム分子量)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、更に水を標線まで加えて1,000mLとする。
ヒ素標準原液 一般試験法のヒ素試験法に定めるところによるものとする。
ヒ素標準液 一般試験法のヒ素試験法に定めるところによるものとする。
フタル酸塩pH標準液 pH標準液、フタル酸塩の項に定める。
フッ素標準液 フッ化ナトリウム(標準試薬)を500~550℃で40~50分間乾燥した後、デシケーター(シリカゲル)で放冷し、その1.105g(1.1045~1.1054g)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。この溶液1mLは、フッ素(F)0.005mgを含む。
ホウ酸塩pH標準液 pH標準液、ホウ酸塩の項に定める。
ホルムアルデヒド標準液 ホルマリン(37%相当)0.54g(0.535~0.544g)を量り、水を入れて溶かし、全量フラスコに入れ、更に水を標線まで加えて1,000mLとする。この溶液10mlを全量ピペットを用いて量り、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。この溶液1mLは、ホルムアルデヒド(HCHO)2μgを含む。用時調製する。
マンガン標準液 過マンガン酸カリウム0.2876g(0.28755~0.28764g)を量り、水100mL及び硫酸1mLを加えて溶かし、亜硫酸水素ナトリウム0.5g(0.45~0.54g)を加え、煮沸し、放冷した後、200mLの全量フラスコに入れ、水を標線まで加えて200mLとし、この溶液20mLを全量ピペットを用いて量り、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。この溶液1mLは、マンガン(Mn)0.01mgを含む。
水・メタノール標準液 一般試験法の水分定量法に定めるところによる。
誘導結合プラズマ質量分析用ゲルマニウム標準液 計量法で規定される標準液。この液1mLはゲルマニウム(Ge)1mgを含む。
誘導結合プラズマ質量分析用鉛標準液 計量法で規定される標準液。この液1mLは鉛(Pb)1mgを含む。
誘導結合プラズマ質量分析用ビスマス標準液 計量法で規定される標準液。この液1mLはビスマス(Bi)1mgを含む。
誘導結合プラズマ質量分析用ヒ素標準液 計量法で規定される標準液。この液1mLはヒ素(As2O3)1.3mgを含む。
リン酸塩pH標準液 pH標準液、リン酸塩の項に定める。
リン酸標準液 リン酸二水素カリウムをデシケーター(シリカゲル)で恒量になるまで乾燥し、その0.3582g(0.35815~0.35824g)を量り、硫酸(3→10)10mLを加え、1,000mLの全量フラスコに入れ、水を標線まで加えて1,000mLとする。この溶液10mLを全量ピペットを用いて量り、100mLの全量フラスコに入れ、水を標線まで加えて100mLとする。この溶液1mLは、リン酸(PO4として)0.025mgを含む。
リン標準液 リン酸二水素カリウムをデシケーター(シリカゲル)で恒量になるまで乾燥し、その10.99g(10.985~10.994g)を量り、水を加えて溶かし、1,000mLの全量フラスコに入れ、更に水を標線まで加えて1,000mLとする。この溶液20mLを全量ピペットを用いて量り、500mLの全量フラスコに入れ、水を標線まで加えて500mLとしてリン標準液とする。この溶液1mLは、リン(P)0.1mgを含む。
(5) 色の比較液
色の比較液は、飼料添加物の試験において、色の比較の対照に用いるものである。
色の比較液は、次の比較原液から調製する。比較原液は、次の方法により調製し、共栓瓶に保存する。色の比較液を用いて液の色を比較するには、別に規定する場合を除き、ネスラー管に入れ、白色の背景を用いて側方から観察する。
塩化コバルトの色の比較原液
塩化コバルト65g(64.5~65.4g)を量り、塩酸25mL及び水を加えて溶かし、1,000mLとする。この溶液5mLを全量ピペットを用いて量り、ヨウ素瓶に入れ、過酸化水素試液5mL及び水酸化ナトリウム溶液(1→5)15mLを加え、10分間煮沸する。放冷した後、ヨウ化カリウム2g(1.5~2.4g)及び硫酸(1→4)20mLを加え、沈殿が溶けた後、遊離したヨウ素を0.1mol/Lチオ硫酸ナトリウム溶液で滴定する(指示薬 デンプン試液1mL)。
0.1mol/Lチオ硫酸ナトリウム溶液1mL=23.79mgCoCl2・6H2O
滴定により得た数値から、1mL中に塩化コバルト(CoCl2・6H2O:237.93)59.48mgを含むように塩酸(1→40)を加えて比較原液とする。
塩化第二鉄の色の比較原液
塩化第二鉄55g(54.5~55.4g)を量り、塩酸25mL及び水を加えて溶かし、1,000mLとする。この溶液10mLを全量ピペットを用いて量り、ヨウ素瓶に入れ、水15mL及びヨウ化カリウム3g(2.5~3.4g)を加え、密栓し、暗所で15分間放置した後、水100mLを加え、遊離したヨウ素を0.1mol/Lチオ硫酸ナトリウム溶液で滴定する(指示薬 デンプン試液1mL)。
0.1mol/Lチオ硫酸ナトリウム溶液1mL=27.03mgFeCl3・6H2O
滴定により得た数値から、1mL中に塩化第二鉄(FeCl3・6H2O:270.30)45.05mgを含むように塩酸(1→40)を加えて比較原液とする。
硫酸銅の色の比較原液
硫酸銅65g(64.5~65.4g)を量り、塩酸25mL及び水を加えて溶かし、1,000mLとする。この溶液10mLを全量ピペットを用いて量り、ヨウ素瓶に入れ、酢酸4mL及びヨウ化カリウム3g(2.5~3.4g)を加え、遊離したヨウ素を0.1mol/Lチオ硫酸ナトリウム溶液で滴定する(指示薬 デンプン試薬1mL)。
0.1mol/Lチオ硫酸ナトリウム溶液1mL=24.97mgCuSO4・5H2O
滴定により得た数値から、1mL中に硫酸銅(CuSO4・5H2O:249.69)62.42mgを含むように塩酸(1→40)を加えて比較原液とする。
色の比較液
次の表に示すそれぞれの色の比較原液及び水の一定量を0.1mL以下の目盛りのあるビュレット又はピペットを用いて量り、混和して調製する。
| 色の比較 液の記号 |
塩化コバルトの 色の比較原液 (mL) |
塩化第二鉄の 色の比較原液 (mL) |
硫酸銅の 色の比較原液 (mL) |
水 (mL) |
| A B C D E F G H I J K L M N O P Q R S T |
0.1 0.3 0.1 0.3 0.4 0.3 0.5 0.2 0.4 0.4 0.5 0.8 0.1 - 0.1 0.2 0.2 0.3 0.2 0.5 |
0.4 0.9 0.6 0.6 1.2 1.2 1.2 1.5 2.2 3.5 4.5 3.8 2.0 4.9 4.8 0.4 0.3 0.4 0.1 0.5 |
0.1 0.3 0.1 0.4 0.3 - 0.2 - 0.1 0.1 - 0.1 0.1 0.1 0.1 0.1 0.1 0.2 - 0.4 |
4.4 3.5 4.2 3.7 3.1 3.5 3.1 3.3 2.3 1.0 - 0.3 2.8 - - 4.3 4.4 4.1 4.7 3.6 |
(6) 計量器・用器
計量器は、飼料添加物の試験において、計量に用いる器具又は機械である。
用器は、飼料添加物の試験において、その条件をできる限り一定にするために定めた器具である。
温度計
通例、浸線付温度計(棒状)又は日本工業規格の全没式水銀温度計(棒状)の器差試験を行ったものを用いる。ただし、凝固点測定法、融点測定法(第1法)、沸点測定法及び蒸留試験法には、浸線付温度計(棒状)を用いる。
浸線付温度計(棒状)は、次に示すものとする。
| 1号 | 2号 | 3号 | 4号 | 5号 | 6号 | |
|---|---|---|---|---|---|---|
| 液体 | 水銀 | 水銀 | 水銀 | 水銀 | 水銀 | 水銀 |
| 液上に満たす気体 | 窒素 | 窒素 | 窒素 | 窒素 | 窒素 | 窒素 |
| 温度範囲 | -17~50℃ | 40~100℃ | 90~150℃ | 140~200℃ | 190~250℃ | 240~320℃ |
| 最小目盛り | 0.2℃ | 0.2℃ | 0.2℃ | 0.2℃ | 0.2℃ | 0.2℃ |
| 長目盛線 | 1℃ごと | 1℃ごと | 1℃ごと | 1℃ごと | 1℃ごと | 1℃ごと |
| 目盛数字 | 2℃ごと | 2℃ごと | 2℃ごと | 2℃ごと | 2℃ごと | 2℃ごと |
| 全長(mm) | 280~300 | 280~300 | 280~300 | 280~300 | 280~300 | 280~300 |
| ミキの直径(mm) | 6.0±0.1 | 6.0±0.1 | 6.0±0.1 | 6.0±0.1 | 6.0±0.1 | 6.0±0.1 |
| 水銀球の長さ(mm) | 12~15 | 12~15 | 12~15 | 12~15 | 12~15 | 12~15 |
| 水銀球の下端から 最低目盛線までの 距離(mm) |
75~90 | 75~90 | 75~90 | 75~90 | 75~90 | 75~90 |
| 温度計の上端から 最高目盛線までの 距離(mm) |
35~50 |
35~50 |
35~50 |
35~50 |
35~50 |
35~50 |
| 水銀球の下端から 浸線までの 距離(mm) |
60 |
60 |
60 |
60 |
60 |
60 |
| 頂部形状 | 環状 | 環状 | 環状 | 環状 | 環状 | 環状 |
| 許容誤差 | 0.2℃ | 0.2℃ | 0.2℃ | 0.2℃ | 0.2℃ | 0.4℃ |
化学用体積計
全量フラスコ、ピペット、ビュレット及びメスシリンダーは、検度したものを用いる。
ネスラー管
無色で、厚さ1.0~1.5mmの硬質ガラス製の共栓付円筒で、図に示すものであって、それぞれの管の50mL目盛線の高さの差が2mm以下のものを用いる。
はかり及び分銅
① 化学はかり 0.1mgまで読み取れるものを用いる。
② セミミクロはかり 0.01mgまで読み取れるものを用いる。
③ ミクロはかり 0.001mgまで読み取れるものを用いる。
④ 分銅 器差試験を行ったものを用いる。
ふるい
日本産業規格の標準ふるいを用いる。
(7) ろ紙
ろ紙は、ろ過の目的に適するように特に留意して作られた紙である。
ろ紙は、次に示す規格のものを用いる。なお、「ろ紙」と記載し、特にその種類を示さないものは、定性分析用ろ紙を示す。ろ紙は、ガス等により汚染されないように保存する。
定性分析用ろ紙 日本産業規格のろ紙(化学分析用)の定性分析用の規格に適合するものとする。
定量分析用ろ紙 日本産業規格のろ紙(化学分析用)の定量分析用の規格に適合するものとする。
クロマトグラフィー用ろ紙 定量分析用のろ紙の規格及び次に示す規格に適合するものとする。ただし、α繊維素含量、銅価、pH、灰分量、ろ水時間及び湿潤破裂強さの試験にあっては、日本産業規格の規定の方法により、吸水高度の試験にあっては、次に示す方法により行う。
吸水高度の試験
| 装置 |  |
| 図に示すものを用いる。 | |
| A1及びA2:ろ紙保持用ガラスブロック | |
| B:三角フラスコ(容量約1,000mL) | |
| C:試料ろ紙 |
操作法
三角フラスコBに水約300mLを入れ、フラスコの口の上にろ紙保持用ガラスブロックA1及びA2を並べて置く。あらかじめ鉛筆で1cmごとに目盛りをつけた試料ろ紙をガラスブロックの間に挟み、初めは静かに滑らせ、ろ紙の下端が水面に着いた後、速やかに滑らせて、目盛りの0点を水面に一致させて固定し、蒸留水が10分間に上昇する高さを測定する。
(8) 滅菌法
滅菌とは、物質中の全ての微生物を殺滅又は除去することをいう。滅菌法は、一般に、微生物の種類、汚染状況、滅菌されるものの性質及び状態に応じて、通例、次に示す方法を単独で又は併用して行う。
滅菌の適否は、通例、無菌試験法により判定する。
滅菌操作は、温度、圧力等が目的とする滅菌条件に適合していることを十分確認して行わなければならない。なお、滅菌条件の選定又は滅菌効果の確認等を行うとき、それぞれの滅菌条件に適した指標菌を用いることができる。
加熱滅菌法
加熱滅菌法を行うとき、温度又は圧力等が規定の滅菌条件に至るまでの加熱時間は、滅菌されるものの性質、容器の大きさ及び収納状態等により異なる。なお、滅菌時間は、滅菌されるものの全ての部分が規定の温度に達してから起算する。
① 火炎滅菌法
火炎中で加熱することにより微生物を殺滅する方法をいう。
本法は、主としてガラス製、磁製又は金属製の物品等で、火炎により破損しないものに用いる。通例、ブンゼンバーナー又はアルコールランプの火炎中で20秒以上加熱する。
② 乾熱滅菌法
乾熱空気中で加熱することにより微生物を殺滅する方法をいう。
本法は、主としてガラス製、磁製、金属製若しくは繊維製の物品、鉱油、脂肪、脂肪油、試薬又は固形の飼料添加物等で乾燥高温に耐えるものに用いる。ガス又は電気により直接加熱する方式、加熱した空気を循環させて乾燥高温状態を保つ方式等があり、通例、次のいずれかの条件で滅菌を行う。
135~145℃ 3~5時間
160~170℃ 2~4時間
180~200℃ 0.5~1時間
200℃以上 0.5時間以上
また、密封容器に入れた飼料添加物の水溶液等で高温に耐えるものは、134~138℃で3分以上乾熱滅菌する方法を用いることができる。
③ 高圧蒸気滅菌法
適当な温度及び圧力の飽和水蒸気中で加熱することにより微生物を殺滅する方法をいう。
本法は、主としてガラス製、磁製、金属製、ゴム製、紙製若しくは繊維製の物品、水、培地、試薬・試液又は液状の飼料添加物等で、高温高圧水蒸気に耐えるものに用いる。滅菌を確実にするために、滅菌器中の空気は、操作中排気口からできる限り排除し、滅菌されるものが飽和水蒸気で満たされるようにしなければならない。通例、次のいずれかの条件で滅菌を行う。
115℃(0.7kg/cm2) 30分間
121℃(1.0kg/cm2) 20分間
126℃(1.4kg/cm2) 15分間
④ 流通蒸気滅菌法
加熱水蒸気を直接流通させることにより微生物を殺滅する方法をいう。
本法は、主としてガラス製、磁製、金属製、ゴム製若しくは繊維製の物品、水、培地、試薬・試液又は液状の飼料添加物等で、乾熱滅菌法又は高圧蒸気滅菌法により変質するおそれのあるものに用いる。通例、100℃の流通水蒸気中で30~60分間滅菌を行う。
⑤ 煮沸滅菌法
沸騰水中に沈め、加熱することにより微生物を殺滅する方法をいう。
本法は、主としてガラス製、磁製、金属製、ゴム製若しくは繊維製の物品、培地、試薬・試液又は液状の飼料添加物等で、乾熱滅菌法又は高圧蒸気滅菌法により変質するおそれがあるものに用いる。なお、殺菌効果を増加するため、沸騰水中に炭酸ナトリウムを1~2%加えることができる。通例、沸騰水中に沈め、15分以上煮沸して滅菌を行う。
⑥ 間けつ滅菌法
80~100℃の水中又は流通水蒸気中で24時間ごとに1回30~60分間ずつ加熱を繰り返すことを、3~5回行うことにより微生物を殺滅する方法をいう。なお、60~80℃で同様に加温を繰り返す低温間けつ滅菌法もある。
本法は、主としてゴム製の物品、培地、試薬・試液又は液状の飼料添加物等で、乾熱滅菌法又は高圧蒸気滅菌法で変質するおそれのあるものに用いる。
ろ過滅菌法
適当なろ過装置を用いてろ過し、微生物を除去する方法をいう。
本法は、主として気体、水、可溶性で熱に不安定な物質を含有する培地、試液又は液状の飼料添加物等に用いる。通例、ろ過装置には、メンブランフィルター、磁製フィルター又はガラスフィルター等が用いられる。
照射滅菌法
① 放射線滅菌法
放射性同位元素を含む線源からのガンマ線を照射することにより微生物を殺滅する方法をいう。
本法は、主としてガラス製、磁製、金属製、ゴム製、プラスチック製又は繊維製の物品等で、放射線照射に耐えるものに用いる。通例、60Co又は137Cs等を含む放射線源が用いられ、滅菌されるものの材質、物理的・化学的性質又は汚染状況等により照射総線量を調節し、滅菌を行う。
② 紫外線滅菌法
紫外線を照射することにより微生物を殺滅する方法をいう。
本法は、主としてガラス製、金属製、ゴム製、プラスチック製若しくは繊維製の物品、施設、設備、水又は飼料添加物等で、紫外線照射に耐えるものに用いる。通例、200~300nmの紫外線が用いられる。
③ 高周波滅菌法
高周波を直接照射し、発生する熱により微生物を殺滅する方法をいう。
本法は、主として水、培地、試液又は液状の飼料添加物で、高周波の照射に耐えるものに用いる。通例、915又は2,450MHzの高周波が用いられる。
化学的滅菌法
① ガス滅菌法
エチレンオキサイド又はホルムアルデヒド等の殺菌性ガスを用いて微生物を殺滅する方法をいう。
本法は、主としてガラス製、磁製、金属製、ゴム製、プラスチック製若しくは繊維製の物品、施設、設備又は粉末の飼料添加物等で、使用するガスにより変質しないものに用いる。なお、温度、湿度、ガス濃度又は時間を調節するために主としてガス滅菌器を用いる。滅菌した後、使用したガスの残留又はその副生成物には、特に注意する。
② 薬液滅菌法
薬液を用いて微生物を殺滅する方法をいう。
本法は、主としてガラス製、磁製、金属製、ゴム製、プラスチック製若しくは繊維製の物品、手指、無菌箱又は無菌設備等で、使用する薬液により変質しないものに用いる。通例、消毒用エタノール、0.1%~1w/v%塩化ベンザルコニウム溶液、クレゾール水、フェノール水又はホルマリン水等が用いられる。
無菌操作法
使用する全ての器具及び材料を、前記各項のいずれかにより滅菌した後、無菌箱又は無菌設備内で無菌的に操作する。
本法は、主として前記各項の滅菌法により滅菌した飼料添加物等の調製、充填又は密封等の操作に用い、操作は、できる限り速やかに行う。
(9) ベルトラン糖類定量表
| 糖類(mg) | 各糖類に相当する銅重量(mg) | 糖類(mg) | 各糖類に相当する銅重量(mg) | ||||||||
| 転化糖 | ブドウ糖 | ガラクトース | 麦芽糖 | 乳糖 | 転化糖 | ブドウ糖 | ガラクトース | 麦芽糖 | 乳糖 | ||
| 10 | 20.6 | 20.4 | 19.3 | 11.2 | 14.4 | 56 | 105.7 | 105.8 | 101.5 | 61.4 | 76.2 |
| 11 | 22.6 | 22.4 | 21.2 | 12.3 | 15.8 | 57 | 107.4 | 107.6 | 103.2 | 62.5 | 77.5 |
| 12 | 24.6 | 24.3 | 23.0 | 13.4 | 17.2 | 58 | 109.2 | 109.3 | 104.9 | 63.5 | 78.8 |
| 13 | 26.5 | 26.3 | 24.9 | 14.5 | 18.6 | 59 | 110.9 | 111.1 | 106.6 | 64.6 | 80.1 |
| 14 | 28.5 | 28.3 | 26.7 | 15.6 | 20.0 | 60 | 112.6 | 112.8 | 108.3 | 65.7 | 81.4 |
| 15 | 30.5 | 30.2 | 28.6 | 16.7 | 21.4 | 61 | 114.3 | 114.5 | 110.0 | 66.8 | 82.7 |
| 16 | 32.5 | 32.2 | 30.5 | 17.8 | 22.8 | 62 | 115.9 | 116.2 | 111.6 | 67.9 | 83.9 |
| 17 | 34.5 | 34.2 | 32.3 | 18.9 | 24.2 | 63 | 117.6 | 117.9 | 113.3 | 68.9 | 85.2 |
| 18 | 36.4 | 36.2 | 34.2 | 20.0 | 25.6 | 64 | 119.2 | 119.6 | 115.0 | 70.0 | 86.5 |
| 19 | 38.4 | 38.1 | 36.0 | 21.1 | 27.0 | 65 | 120.9 | 121.3 | 116.6 | 71.1 | 87.7 |
| 20 | 40.4 | 40.1 | 37.9 | 22.2 | 28.4 | 66 | 122.6 | 123.0 | 118.3 | 72.2 | 89.0 |
| 21 | 42.3 | 42.0 | 39.7 | 23.3 | 29.8 | 67 | 124.2 | 124.7 | 120.0 | 73.3 | 90.3 |
| 22 | 44.2 | 43.9 | 41.6 | 24.4 | 31.1 | 68 | 125.9 | 126.4 | 121.7 | 74.3 | 91.6 |
| 23 | 46.1 | 45.8 | 43.4 | 25.5 | 32.5 | 69 | 127.5 | 128.1 | 123.3 | 75.4 | 92.8 |
| 24 | 48.0 | 47.7 | 45.2 | 26.6 | 33.9 | 70 | 129.2 | 129.8 | 125.0 | 76.5 | 94.1 |
| 25 | 49.8 | 49.6 | 47.0 | 27.7 | 35.2 | 71 | 130.8 | 131.4 | 126.6 | 77.6 | 95.4 |
| 26 | 51.7 | 51.5 | 48.9 | 28.9 | 36.6 | 72 | 132.4 | 133.1 | 128.3 | 78.6 | 96.9 |
| 27 | 53.6 | 53.4 | 50.7 | 30.0 | 38.0 | 73 | 134.0 | 134.7 | 130.0 | 79.7 | 98.0 |
| 28 | 55.5 | 55.3 | 52.5 | 31.1 | 39.4 | 74 | 135.6 | 136.3 | 131.5 | 80.8 | 99.1 |
| 29 | 57.4 | 57.2 | 54.4 | 32.2 | 40.7 | 75 | 137.2 | 137.9 | 133.1 | 81.8 | 100.4 |
| 30 | 59.3 | 59.1 | 56.2 | 33.3 | 42.1 | 76 | 138.9 | 139.6 | 134.8 | 82.9 | 101.7 |
| 31 | 61.1 | 60.9 | 58.0 | 34.4 | 43.4 | 77 | 140.5 | 141.2 | 136.4 | 84.0 | 102.9 |
| 32 | 63.0 | 62.8 | 59.7 | 35.5 | 44.8 | 78 | 142.1 | 142.8 | 138.0 | 85.1 | 104.2 |
| 33 | 64.8 | 64.6 | 61.5 | 36.5 | 46.1 | 79 | 143.7 | 144.5 | 139.7 | 86.1 | 105.4 |
| 34 | 66.7 | 66.5 | 63.3 | 37.6 | 47.4 | 80 | 145.3 | 146.1 | 141.3 | 87.2 | 106.7 |
| 35 | 68.5 | 68.3 | 65.0 | 38.7 | 48.7 | 81 | 146.9 | 147.7 | 142.9 | 88.3 | 107.9 |
| 36 | 70.3 | 70.1 | 66.8 | 39.8 | 50.1 | 82 | 148.5 | 149.3 | 144.6 | 89.4 | 109.2 |
| 37 | 72.2 | 72.0 | 68.6 | 40.9 | 51.4 | 83 | 150.0 | 150.9 | 146.2 | 90.4 | 110.4 |
| 38 | 74.0 | 73.8 | 70.4 | 41.9 | 52.7 | 84 | 151.6 | 152.5 | 147.8 | 91.5 | 111.7 |
| 39 | 75.9 | 75.7 | 72.1 | 43.0 | 54.1 | 85 | 153.2 | 154.0 | 149.4 | 92.6 | 112.9 |
| 40 | 77.7 | 77.5 | 73.9 | 44.1 | 55.4 | 86 | 154.8 | 155.6 | 151.1 | 93.7 | 114.1 |
| 41 | 79.5 | 79.3 | 75.6 | 45.2 | 56.7 | 87 | 156.4 | 157.2 | 152.7 | 94.8 | 115.4 |
| 42 | 81.2 | 81.1 | 77.4 | 46.3 | 58.0 | 88 | 157.9 | 158.8 | 154.3 | 95.8 | 116.6 |
| 43 | 83.0 | 82.9 | 79.1 | 47.4 | 59.3 | 89 | 159.5 | 160.4 | 156.0 | 96.9 | 117.9 |
| 44 | 84.8 | 84.7 | 80.8 | 48.5 | 60.6 | 90 | 161.1 | 162.0 | 157.6 | 98.0 | 119.1 |
| 45 | 86.5 | 86.4 | 82.5 | 49.5 | 61.9 | 91 | 162.6 | 163.6 | 159.2 | 99.0 | 120.3 |
| 46 | 88.3 | 88.2 | 84.3 | 50.6 | 63.3 | 92 | 164.2 | 165.2 | 160.8 | 100.1 | 121.6 |
| 47 | 90.1 | 90.0 | 86.0 | 51.7 | 64.6 | 93 | 165.7 | 166.7 | 162.4 | 101.1 | 122.8 |
| 48 | 91.9 | 91.8 | 87.7 | 52.8 | 65.9 | 94 | 167.3 | 168.3 | 164.0 | 102.2 | 124.0 |
| 49 | 93.6 | 93.6 | 89.5 | 53.9 | 67.2 | 95 | 168.8 | 169.9 | 165.6 | 103.2 | 125.2 |
| 50 | 95.4 | 95.4 | 91.2 | 55.0 | 68.5 | 96 | 170.3 | 171.5 | 167.2 | 104.2 | 126.5 |
| 51 | 97.1 | 97.1 | 92.9 | 56.1 | 69.8 | 97 | 171.9 | 173.1 | 168.8 | 105.3 | 127.7 |
| 52 | 98.8 | 98.9 | 94.6 | 57.1 | 71.1 | 98 | 173.4 | 174.6 | 170.4 | 106.3 | 128.9 |
| 53 | 100.6 | 100.6 | 96.3 | 58.2 | 72.4 | 99 | 175.0 | 176.2 | 172.0 | 107.4 | 130.2 |
| 54 | 102.2 | 102.3 | 98.0 | 59.3 | 73.7 | 100 | 176.5 | 177.8 | 173.6 | 108.4 | 131.4 |
| 55 | 104.0 | 104.1 | 99.7 | 60.3 | 74.9 | ||||||
8 各飼料添加物の成分規格及び製造の方法等の基準 (略)
附 則
1 この省令は、飼料の品質改善に関する法律の一部を改正する法律(昭和50年法律第68号)の施行の日(昭和51年7月24日)から施行する。
2 この省令の施行の日から6月間は、法第2条の3各号に掲げる行為については、同条の規定は、適用しない。
3 この省令の施行の日から6月間に製造業者、輸入業者又は販売業者が法第2条の3第2号から第4号までの規定に規定する飼料又は飼料添加物を販売した場合における当該飼料又は当該飼料添加物については、法第2条の7(第1号に係る部分に限る。)の規定は、適用しない。
4 法第2条の3各号に掲げる行為であつてぎんざけに使用される飼料又は当該飼料に用いられる飼料添加物(ぎんざけ以外の飼料の安全性の確保及び品質の改善に関する法律施行令(昭和51年政令第198号)第1条に規定する動物にも使用される飼料又は当該飼料にも用いられる飼料添加物を除く。以下「追加飼料等」という。)に係るものについては、飼料の安全性の確保及び品質の改善に関する法律施行令の一部を改正する政令(平成2年政令第199号)の施行の日から6月間は、法第2条の3の規定は、適用しない。
5 前項に規定する期間内に追加飼料等の製造業者、輸入業者又は販売業者が追加飼料等で法第2条の3第2号から第4号までに規定する飼料又は飼料添加物に該当するものを販売した場合における当該追加飼料等については、法第2条の7(第1号に係る部分に限る。)の規定は、適用しない。
附 則(昭和53年7月5日農林省令第49号) 抄
第1条 この省令は、公布の日から施行する。
附 則(昭和53年9月5日農林水産省令第8号) 抄
1 この省令は、公布の日から施行する。ただし、別表第2の5の改正規定((23)を(60)とし、(15)から(22)までを(52)から(59)までとし、(3)から(14)までを削り、(2)の次に次のように加える部分に限る。)は、昭和54年2月1日から施行する。
附 則(昭和54年11月19日農林水産省令第47号)
1 この省令は、公布の日から施行する。ただし、別表第2の7の改正規定(別表第2の7の(1)に係る部分を除く。)は、昭和55年4月1日から施行する。
2 この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令(以下「改正後の省令」という。)別表第2の7の(8)、(11)、(16)、(23)、(26)、(27)、(29)、(30)、(34)、(61)、(62)、(64)から(68)まで、(98)、(101)及び(102)に規定する飼料添加物又は当該飼料添加物を含む飼料に係る改正後の省令別表第1の1の(5)のイ又は改正後の省令別表第2の4の(2)に規定する事項の記載については、昭和55年3月31日までは、なお従前の例によることができる。
附 則(昭和56年7月27日農林水産省令第31号)
1 この省令は、公布の日から施行する。
2 昭和56年12月31日以前に製造された液状の飼料添加物については、この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令(以下「改正後の省令」という。)別表第2の2の(4)の規定にかかわらず、なお従前の例によることができる。
3 改正後の省令別表第2の7の(74)から(96)までに規定する飼料添加物の成分規格及び製造の方法等の基準については、昭和56年12月31日までは、なお従前の例によることができる。
4 改正後の省令別表第2の7の(1)、(2)、(3)、(75)、(77)、(78)、(81)及び(89)に規定する飼料添加物を含む飼料に係る改正後の省令別表第1の1の(5)のイに規定する事項の記載については、昭和56年12月31日までは、なお従前の例によることができる。
5 改正後の省令別表第2の7の(1)、(2)、(3)、(75)、(77)、(78)、(81)及び(89)に規定する飼料添加物に係る改正後の省令別表第2の4の(2)に規定する事項の記載については、昭和56年12月31日までは、なお従前の例によることができる。
附 則(昭和58年7月6日農林水産省令第23号)
1 この省令は、公布の日から施行する。ただし、第2条の改正規定は、昭和59年1月1日から施行する。
2 この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令の別表第1の1の(1)のクロピドール及びナイカルバジンに係る飼料1般の成分規格並びに別表第2の7の(74)、(75)、(89)、(91)及び(95)に規定する表示の基準については、昭和58年12月31日までは、なお従前の例によることができる。
附 則(昭和60年10月15日農林水産省令第47号)
1 この省令は、公布の日から施行する。ただし、第2条の改正規定は、昭和61年4月1日から施行する。
2 第1条の改正規定による改正後の飼料及び飼料添加物の成分規格等に関する省令の別表第1の1の(1)のチオペプチンに係る飼料1般の成分規格並びに別表第2の7の(1)、(3)、(4)、(5)、(7)、(8)、(14)、(15)、(90)、(100)、(113)及び(114)の規定に規定する飼料添加物又は同7の(1)、(3)、(4)、(5)、(7)、(8)、(14)、(15)、(113)及び(114)の規定に規定する飼料添加物を含む飼料に係る改正後の省令別表第1の1の(5)のイ又は改正後の省令別表第2の4の(2)に規定する事項の記載については、昭和61年3月31日までは、なお従前の例によることができる。
附 則(昭和62年12月25日農林水産省令第46号)
この省令は、公布の日から施行する。
附 則(平成2年3月20日農林水産省令第7号)
1 この省令は、公布の日から施行する。ただし、第2条の規定は、平成2年9月1日から施行する。
2 飼料及び飼料添加物の成分規格等に関する省令別表第1の(1)の表に掲げる対象飼料が含むことができるアルキルトリメチルアンモニウムカルシウムオキシテトラサイクリン、キタサマイシン、クロルテトラサイクリン、デストマイシンA、ハイグロマイシンB、フラボフォスフォリポール、硫酸コリスチン及びリン酸タイロシンの量については、平成2年8月31日までは、第1条の規定による改正後の同令別表第1の1の(1)のイの規定にかかわらず、なお従前の例によることができる。
3 ポリオキシエチレングリセリン脂肪酸エステル、アミラーゼ、アルカリ性プロテアーゼ、キシラナーゼ・ペクチナーゼ複合酵素、酸性プロテアーゼ、セルラーゼ、セルラーゼ・プロテアーゼ・ペクチナーゼ複合酵素、中性プロテアーゼ、ラクターゼ若しくはリパーゼ又はこれらを含む飼料の表示については、平成2年8月31日までは、第1条の規定による改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(5)のイ並びに別表第2の4の(2)及び7の(117)から(125)までの規定にかかわらず、なお従前の例によることができる。
附 則(平成2年6月29日農林水産省令第30号)
この省令は、飼料の安全性の確保及び品質の改善に関する法律施行令の一部を改正する政令の施行の日から施行する。
附 則(平成3年6月3日農林水産省令第29号)
1 この省令は、公布の日から施行する。
2 飼料及び飼料添加物の成分規格等に関する省令(以下「成分規格等省令」という。)別表第1の(1)の表に掲げる対象飼料が含むことができるサリノマイシンナトリウム及びリン酸タイロシンの量については、平成3年11月3日までは、この省令による改正後の成分規格等省令別表第1の1の(1)のイの規定にかかわらず、なお従前の例によることができる。
3 モネンシンナトリウム及びラサロシドナトリウムの表示については、平成3年11月3日までは、改正後の成分規格等省令別表第2の4の(2)並びに7の(107)及び(108)の規定にかかわらず、なお従前の例によることができる。
附 則(平成4年5月14日農林水産省令第28号)
1 この省令は、公布の日から施行する。
2 ギ酸又はこれらを含む飼料の表示については、平成4年10月31日までは改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(5)のイ並びに別表第2の4の(2)及び7の(5)の規定にかかわらず、なお従前の例によることができる。
附 則(平成5年6月22日農林水産省令第28号)
1 この省令は、公布の日から施行する。
2 改正後の飼料及び飼料添加物の成分規格等に関する省令別表第2の7の(95)に規定するアボパルシンの成分規格及び製造の方法の基準については、平成5年11月3日までは、なお従前の例によることができる。
附 則(平成5年12月20日農林水産省令第66号)
この省令は、平成6年6月1日から施行する。
附 則(平成6年7月18日農林水産省令第45号)
1 この省令は、公布の日から施行する。
2 改正後の飼料及び飼料添加物の成分規格等に関する省令別表第2の7の(119)のイの(ア)に規定するオラキンドックスの成分規格については、平成6年12月31日までは、なお従前の例によることができる。
附 則(平成7年8月28日農林水産省令第48号)
1 この省令は、公布の日から施行する。
2 次の各号のいずれかに該当する飼料添加物又は飼料の表示については、平成8年1月31日までは、改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(5)のイ及び別表第2の4の(2)の規定に関わらず、なお、従前の例によることができる。
1 エンテロコッカス フェカーリス、エンテロコッカス フェシウム、クロストリジウム ブチリカム、バチルス コアグランス、バチルス サブチルス、バチルス セレウス、ビフィドバクテリウム サーモフィラム、ビフィドバクテリウム シュードロンガム、ラクトバチルス アシドフィルス及びラクトバチルス サリバリウス並びにこれらのいずれかを有効成分として含有する飼料添加物
2 飼料添加物を含む飼料
附 則(平成8年5月17日農林水産省令第23号)
この省令は、公布の日から施行する。
附 則(平成8年9月20日農林水産省令第51号)
1 この省令は、公布の日から施行する。
2 飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(1)のイの表に掲げる対象飼料が含むことができるバージニアマイシンの量については、平成9年3月19日までは、この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(1)のイの規定にかかわらず、なお従前の例によることができる。
附 則(平成9年3月18日農林水産省令第11号)
この省令は、公布の日から施行する。
附 則(平成9年9月30日農林水産省令第69号)
この省令は、公布の日から施行する。
附 則(平成10年7月6日農林水産省令第57号)
この省令は、公布の日から施行する。
附 則(平成10年8月21日農林水産省令第66号)
この省令は、公布の日から施行する。
附 則(平成11年12月22日農林水産省令第88号)
この省令は、公布の日から施行する。
附 則(平成12年7月17日農林水産省令第75号)
この省令は、公布の日から施行する。
附 則(平成13年3月7日農林水産省令第51号)
この省令は、公布の日から施行する。
附 則(平成13年3月7日農林水産省令第52号)
この省令は、平成13年4月1日から施行する。
附 則(平成13年3月22日農林水産省令第59号) 抄
(施行期日)
第1条 この省令は、平成13年4月1日から施行する。
(処分、申請等に関する経過措置)
第3条 この省令の施行前に改正前のそれぞれの省令の規定によりされた承認等の処分その他の行為(以下「承認等の行為」という。)又はこの省令の施行の際現に改正前のそれぞれの省令の規定によりされている承認等の申請その他の行為(以下「申請等の行為」という。)は、この省令の施行の日以後における改正後のそれぞれの省令の適用については、改正後のそれぞれの省令の相当規定によりされた承認等の行為又は申請等の行為とみなす。
附 則(平成13年9月18日農林水産省令第123号)
この省令は、公布の日から施行する。ただし、第2条の規定は、平成14年1月1日から施行する。
附 則(平成13年10月15日農林水産省令第133号)
1 この省令は、公布の日から施行する。ただし、別表第1の1の(5)のイ中「、反すう動物等由来たん白質を含むもの」を削り、同イの(サ)を削る改正規定は、平成14年1月1日から施行する。
2 この省令の施行の日以前に飼料の製造業者が販売した飼料については、この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(1)のケからサまで、同(2)のキ及び同(3)のカの規定にかかわらず、平成13年10月31日までは、なお従前の例によることができる。
附 則(平成13年11月1日農林水産省令第137号)
この省令は、公布の日から施行する。ただし、別表第1の1の(5)の改正規定は、平成14年1月1日から施行する。
附 則(平成13年12月19日農林水産省令第146号)
この省令は、公布の日から施行する。
附 則(平成14年4月25日農林水産省令第40号)
この省令は、公布の日から施行する。
附 則(平成14年8月2日農林水産省令第70号)
この省令は、公布の日から施行する。
附 則(平成14年11月26日農林水産省令第88号)
1 この省令は、平成15年4月1日から施行する。
2 この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(1)のチ及びツ、同表の1の(2)のシ、別表第2の2並びに同表の3の(7)に規定する確認は、この省令の施行前においても行うことができる。
附 則(平成15年5月26日農林水産省令第50号)
この省令は、公布の日から施行する。
附 則(平成15年6月27日農林水産省令第64号)
(施行期日)
第1条 この省令は、飼料の安全性の確保及び品質の改善に関する法律の一部を改正する等の法律の施行の日(平成15年7月1日)から施行する。
(経過措置)
第2条 牛用の飼料又は当該飼料に用いられる飼料添加物をめん羊、山羊及びしかに使用する場合には、この省令の施行の日から2年間は、法第4条第1号及び第4号(使用に係る部分に限る。)の規定は、適用しない。
2 この省令の施行の際現に牛、めん羊、山羊又はしかを対象とする飼料(飼料を製造するための原料又は材料を含む。)をほ乳動物由来たん白質、家きん由来たん白質又は魚介類由来たん白質を含む飼料(飼料を製造するための原料又は材料を含む。)の製造工程と同1の製造工程において製造している飼料の製造業者については、この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(2)のスの規定は、平成17年3月31日までは、適用しない。
3 確認済血粉等若しくは確認済チキンミール等又はこれらを原料とする飼料に係る表示については、平成15年12月31日までは、この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(5)のイの(サ)の規定にかかわらず、なお従前の例によることができる。
附 則(平成15年6月30日農林水産省令第67号)
1 この省令は、平成16年1月1日から施行する。
2 この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(1)のセに規定する確認は、この省令の施行前においても行うことができる。
附 則(平成16年1月15日農林水産省令第4号)
1 この省令は、平成16年5月1日から施行する。
2 この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令(以下「新令」という。)別表第1の4の(1)のウに規定する確認は、この省令の施行前においても行うことができる。
3 この省令の施行前に製造された飼料については、新令別表第1の4の規定にかかわらず、平成16年6月30日までは、なお従前の例によることができる。
附 則(平成16年10月12日農林水産省令第79号)
この省令は、公布の日から施行する。
附 則(平成16年10月27日農林水産省令第82号)
この省令は、平成17年2月1日から施行する。
附 則(平成17年2月28日農林水産省令第15号)
この省令は、平成17年4月1日から施行する。
附 則(平成18年5月22日農林水産省令第49号)
この省令は、平成18年5月29日から施行する。
附 則(平成18年9月1日農林水産省令第74号)
この省令は、公布の日から施行する。
附 則(平成19年3月30日農林水産省令第28号)
(施行期日)
第1条 この省令は、平成19年4月1日から施行する。
附 則(平成20年8月29日農林水産省令第55号)
1 この省令は、公布の日から施行する。
2 飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(1)のウの表に掲げる対象飼料が含むことができるアビラマイシンの量、別表第2の8の(139)のフィターゼ(その2の(2))のイの(ウ)の有効期間及び別表第2の8の(139)のフィターゼ(その2の(2))のウの(ウ)の有効期間については、平成21年2月28日までは、この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の1の(1)のウの規定、別表第2の8の(139)のフィターゼ(その2)のイの(ウ)の規定及び別表第2の8の(139)のフィターゼ(その2の(2))のウの(ウ)の規定にかかわらず、なお従前の例によることができる。
附 則(平成20年11月14日農林水産省令第72号)
この省令は、公布の日から施行する。
附 則(平成21年6月23日農林水産省令第40号)
この省令は、公布の日から施行する。
附 則(平成22年2月4日農林水産省令第9号)
この省令は、公布の日から施行する。
附 則(平成22年5月31日農林水産省令第40号)
この省令は、公布の日から施行する。
附 則(平成24年11月22日農林水産省令第57号)
この省令は、平成25年5月22日から施行する。
附 則(平成25年3月25日農林水産省令第17号)
この省令は、平成25年4月1日から施行する。
附則(平成26年2月6日農林水産省令第6号)
この省令は、公布の日から施行する。
附則(平成26年6月11日農林水産省令第36号)
この省令は、公布の日から施行する。
附則(平成26年7月23日農林水産省令第44号)
この省令は、公布の日から施行する。
附則(平成27年3月26日農林水産省令第17号)
この省令は、平成27年4月1日から施行する。
附則(平成27年7月6日農林水産省令第63号)
この省令は、公布の日から施行する。
附則(平成27年7月27日農林水産省令第65号)
この省令は、公布の日から施行する。ただし、第2条の規定は、公布の日から起算して6月を経過した日から施行する。
附則(平成27年11月26日農林水産省令第81号)
この省令は、公布の日から施行する。
附則(平成27年12月7日農林水産省令第82号)
この省令は、公布の日から施行する。
附則(平成27年12月18日農林水産省令第84号)
この省令は、公布の日から施行する。ただし、第2条の規定は、公布の日から起算して6月を経過した日から施行する。
附則(平成28年3月23日農林水産省令第15号)
この省令は、公布の日から施行する。
附則(平成28年4月18日農林水産省令第33号)
この省令は、公布の日から施行する。
附則(平成29年1月26日農林水産省令第7号)
この省令は、公布の日から施行する。
附則(平成29年12月28日農林水産省令第70号)
この省令は、平成30年7月1日から施行する。ただし、別表第2の8の(112)の改正規定は、公布の日から施行する。
附則(平成30年4月2日農林水産省令第25号)
この省令は、公布の日から施行する。
附則(平成30年7月2日農林水産省令第43号)
この省令は、公布の日から施行する。
附則(平成30年10月19日農林水産省令第69号)
この省令は、公布の日から施行する。
附則(平成30年11月30日農林水産省令第75号)抄
(施行期日)
1 この省令は、農薬取締法の一部を改正する法律の施行の日(平成30年12月1日)から施行する。
(経過措置)
2 (略)
附則(平成30年12月27日農林水産省令第82号)
この省令は、公布の日から施行する。
附則(平成31年4月22日農林水産省令第37号)
この省令は、平成31年5月1日から施行する。
附則(令和元年5月17日農林水産省令第2号)
この省令は、公布の日から施行する。ただし、第2条の規定は、公布の日から起算して6月を経過した日から施行する。
附則(令和元年5月31日農林水産省令第6号)
この省令は、公布の日から施行する。
附則(令和元年6月27日農林水産省令第10号)
(施行期日)
第1条 この省令は、不正競争防止法等の一部を改正する法律の施行の日(令和元年7月1日)から施行する。
(経過措置)
第2条 この省令の施行の際現にある改正前の様式(次項において「旧様式」という。)により使用されている書類は、この省令による改正後の様式によるものとみなす。
2 この省令の施行の際現にある旧様式による用紙については、当分の間、これを取り繕って使用することができる。
附則(令和元年10月8日農林水産省令第36号)
この省令は、公布の日から施行する。ただし、第2条の規定は、令和元年12月27日から施行する。
附則(令和2年1月30日農林水産省令第4号)
この省令は、公布の日から施行する。
附則(令和2年5月28日農林水産省令第38号)
この省令は、公布の日から施行する。
附則(令和2年5月29日農林水産省令第39号)
この省令は、公布の日から施行する。
附則(令和2年6月1日農林水産省令第40号)
(施行期日)
1 この省令は、令和2年12月1日から施行する。
(経過措置)
2 確認済豚血粉等、確認済豚肉骨粉等、確認済馬肉骨粉等、確認済チキンミール等、確認済家きん加水分解たん白質等、確認済魚介類由来たん白質若しくは確認済原料混合肉骨粉等又はこれらを原料とする飼料(確認済牛血粉等又は確認済牛肉骨粉等を含む飼料を除く。)、確認済牛血粉等、確認済牛肉骨粉等又は飼料及び飼料添加物の成分規格等に関する省令別表第1の2の(2)のウの確認を受けた工程で製造された養殖水産動物を対象とする飼料及び確認済動物性油脂(反すう動物由来動物性油脂を含むものに限る。)を含む飼料に係る表示については、令和3年11月30日までは、この省令による改正後の飼料及び飼料添加物の成分規格等に関する省令別表第1の2の(5)のイ及びウ並びに別表第1の5の(5)のオの規定にかかわらず、なお従前の例によることができる。
附則(令和2年8月26日農林水産省令第56号)
この省令は、令和3年4月1日から施行する。
附則(令和2年10月15日農林水産省令第71号)
この省令は、公布の日から施行する。ただし、第2条の規定は、公布の日から起算して6月を経過した日から施行する。
附則(令和2年10月22日農林水産省令第74号)
この省令は、公布の日から施行する。
附則(令和3年3月9日農林水産省令第8号)
この省令は、公布の日から施行する。
附則(令和3年4月15日農林水産省令第30号)
この省令は、公布の日から施行する。
附則(令和4年1月21日農林水産省令第4号)
この省令は、公布の日から施行する。
附則(令和4年10月17日農林水産省令第59号)
この省令は、公布の日から施行する。
附則(令和4年12月6日農林水産省令第71号)
この省令は、公布の日から施行する。
附則(令和5年2月1日農林水産省令第6号)
この省令は、公布の日から施行する。ただし、第2条の規定は、公布の日から起算して6月を経過した日から施行する。
附則(令和5年4月4日農林水産省令第28号)
この省令は、公布の日から施行する。
附則(令和5年4月28日農林水産省令第30号)
この省令は、公布の日から施行する。ただし、第2条の規定は、公布の日から起算して6月を経過した日から施行する。
附則(令和5年7月24日農林水産省令第40号)
この省令は、公布の日から施行する。
附則(令和5年9月26日農林水産省令第47号)
この省令は、公布の日から施行する。
附則(令和6年1月29日農林水産省令第3号)
この省令は、公布の日から施行する。
附則(令和6年3月28日農林水産省令第14号)
この省令は、公布の日から施行する。
附則(令和6年8月26日農林水産省令第44号)
この省令は、公布の日から施行する。
附則(令和6年10月3日農林水産省令第52号)
この省令は、公布の日から施行する。
附則(令和6年11月1日農林水産省令第56号)
この省令は、公布の日から施行する。
附則(令和6年12月25日農林水産省令第64号)
この省令は、公布の日から施行する。
附則(令和7年3月6日農林水産省令第7号)
この省令は、公布の日から施行する。
附則(令和7年5月1日農林水産省令第23号)
この省令は、公布の日から施行する。
附則(令和7年6月10日農林水産省令第27号)
この省令は、公布の日から施行する。
附則(令和7年8月25日農林水産省令第38号)
この省令は、公布の日から施行する。
附則(令和7年12月1日農林水産省令第51号)
この省令は、公布の日から施行する。
附則(令和8年1月6日農林水産省令第1号)
この省令は、公布の日から施行する。 ただし、次の各号に揚げる規定は、当該各号に定める日から施行する。
1 第二条の規定 公布の日から起算して六月を経過した日
2 第三条の規定 公布の日から起算して一年を経過した日
附則(令和8年3月30日農林水産省令第22号)
この省令は、公布の日から施行する。
附則(令和8年5月26日農林水産省令第42号)
この省令は、公布の日から施行する。




