5.21 ニッケル
5.21.1 ジメチルグリオキシム法
A 適用範囲
ケイ酸質肥料などのスラグ類を主な対象とするが,その他広範囲の試料にも適用できる。
B 装置
吸光光度分析装置
C 試薬液の調製
1)標準ニッケル液
特級ニッケル液(Ni)1gをトールビーカーに正確にとり,少量の硝酸で溶かしたのち,硫酸(1+1)5mlを加え加熱して硫酸の白煙を発生させ,冷却後水を加えて正確に1000mlとし,標準ニッケル原液を作成する(この液1mlはNiとして1mgを含有する)。使用に際してこの原液の一定量を水で正確に50倍に希釈する(この液1mlはNiとして20μgを含有する)。
2)ジメチルグリオキシム液
ジメチルグリオキシム(C4H8N2O2)1gをアルコール100mlに溶かす。
3)酒石酸液
酒石酸(C4H6O6)350gを水に溶かして1000mlとする。
D 試料液の調製
5.1.1.D.aによる。
E 定量
試料液10~20ml(Niとして10~500μgがよい)を小型分液漏斗に正確にとり,酒石酸液3mlとリトマス試験紙の小片とを加え,アンモニア水で中和し更にその数滴を過剰に加えたのち,ジメチルグリオキシム液1ml及びクロロホルム5mlを加え約30秒間振り混ぜてしばらく静置する。ニッケルジメチルグリオキシムはクロロホルムに溶けるので下層のクロロホルムを別の分液漏斗に移し,残液には更にクロロホルム3mlを加え,同様の操作を2回繰り返してニッケル塩をことごとくクロロホルムに移行させる。次に分液漏斗に集めたクロロホルムに塩酸(1+20)5mlを加え約30秒間振り混ぜて静置する。ニッケルは塩酸に溶けるので下層のクロロホルムを別の分液漏斗に移し,これに再び塩酸(1+20)5mlを加え,同様の操作を繰り返してニッケルをことごとく塩酸に溶かし,これらの塩酸液を100mlのメスフラスコに合わせ移す。これに塩酸(1+1)5ml及び飽和臭素水5mlを加えて振り混ぜたのち,約10分間静置してニッケルを完全に酸化させる。次にアンモニア水で中和し更にその1mlを過剰に加え,水で約90mlに希釈したのちジメチルグリオキシム液1mlを加えて振り混ぜ,ニッケルジメチルグリオキシムの赤褐色を発色させ冷却後標線まで水を加え,約30分間放置後波長450nm付近の吸光度を測定する。同時に標準ニッケル液を数段階に正確にとり,試料液の場合と同一条件で操作して作成した検量線からニッケル(Ni)の量を求める。
(付記)
微量のクロロホルムにより混濁を生じて吸光度の測定に差し支えのある場合には,乾燥ろ紙でろ過した液を用いる。
5.21.2 原子吸光測光法
A 適用範囲
広範囲の試料に適用される。高含有量の試料では酸分解液を直接噴霧させて測定するが,妨害する成分が存在する場合あるいは微量の場合には溶媒抽出と組み合わすのがよい。
B 装置
原子吸光分析装置 ニッケル中空陰極ランプ。フレームレス原子化装置付きの装置でもよい。
C 試薬液の調製
1)標準ニッケル液
5.21.1.C.1)により作成した標準ニッケル原液の一定量を使用に際して水で正確に500倍に希釈する(この液1mlはNiとして2μgを含有する)。
2)ジエチルジチオカルバミン酸ナトリウム液
ジエチルジチオカルバミン酸ナトリウム[Na(C2H5)2NCS2・3H2O]1gを水に溶かして100mlとする。
3)クエン酸二アンモニウム液
クエン酸二アンモニウム[(NH4)2HC6H5O7]200gを水に溶かして1000mlとする。
4)干渉抑制剤液
4.5.1.2.C.2)による。
D 試料液の調製
5.1.2.D.aによる。
E 定量
試料液の一定量(Niとして50~500μgがよい)を50mlのメスフラスコに正確にとり,干渉抑制剤液5ml及び5M塩酸5mlを加え標線まで水を加える。この液につき原子吸光分析装置により波長232.0nmの吸光度を測定する。同時に標準ニッケル液を数段階にメスフラスコに正確にとり,試料液と同じ濃度となるように5M塩酸及び干渉抑制剤液を加え標線まで水を加えた液につき,試料液の場合と同一条件で測光して作成した検量線からニッケル(Ni)の量を求める。
全操作にわたって空試験を行い,結果を補正する。
(付記)
1.カルシウムなどを多量に含有し定量の妨げとなる場合には,5.6.1.E.aに準じて干渉補償液を標準液に加えるが,バックグラウンドの吸収を別に補償する方法をとらなければならない。
2.ニッケルが微量であり,また共存物質による妨害が上記の方法によってもなおみられる場合には,次の溶媒抽出を行ったのち原子吸光測光するとよい。
試料液の一定量(Niとして1~50μgがよく,液量40ml以下)を小型ビーカーに正確にとり,クエン酸二アンモニウム液5mlを加え,アンモニア水(1+1)を滴下してpHを9~9.5に調製したのち,少量の水で小型分液漏斗に移し,ジエチルジチオカルバミン酸ナトリウム液1mlを加え,更に水を加えて約50mlとする。これに4-メチル-2-ペンタノン(イソブチルメチルケトン)10mlを正確に加え,約1分間激しく振り混ぜて少時静置したのち,4-メチル-2-ペンタノン相をとり原子吸光測光する。
5.22 バナジウム
5.22.1 ピリジルアゾレソルシノール法
A 適用範囲
肥料及びリン鉱石を対象とする。
B 装置
吸光光度分析装置
C 試薬液の調製
1)標準バナジウム液
特級メタバナジン酸アンモニウム(NH4VO3)2.296gを水に溶かして正確に1000mlとし標準バナジウム原液を作成する(この液1mlはVとして1mgを含有する)。使用に際してこの原液の一定量を水で正確に1000倍に希釈する(この液1mlはVとして1μgを含有する)。
2)ピリジルアゾレソルシノール液
4-(2-ピリジルアゾ)レソルシノール(C11H9N3O2)0.2gに水酸化ナトリウム液6.8mlを加えて溶かし,水を加えて約200mlとする。この液に,リン酸一水素二ナトリウム(Na2HPO4・12H2O)30gとリン酸二水素一カリウム(KH2PO4)23gを水約700mlに溶かした液(pH6.4)を加え,更に水を加えて1000mlとする。冷暗所に保存すれば約2か月間使用することができる。
3)1-ブタノール-ベンゼン液
1-ブタノール1容とベンゼン2容とを混合する。
4)水酸化ナトリウム液
水酸化ナトリウム10g水に溶かして1000mlとする。
5)指示薬
メチルオレンジ 5.20.1.C.4).ⅲ)による。
D 試料液の調製
分析試料1~5gをトールビーカーに正確にとり,塩酸約30ml及び硝酸約10mlを加えて時計皿で覆い徐々に加熱し30分間煮沸する。時計皿を除き更に加熱を続けて液量が約10mlになるまで蒸発する。ビーカーの内壁を少量の水で洗浄し,塩酸約15ml及び硝酸約5mlを加え再び加熱して乾固近くまで蒸発し放冷する。適量の水を加え少時加熱して溶かし,冷却後メチルオレンジを指示薬として加え水酸化ナトリウム液で中和し,水で100~500mlのメスフラスコに移し,標線まで水を加えて乾燥ろ紙でろ過する。
E 定量
試料液の一定量(Vとして1~20μgがよい)を小型分液漏斗に正確にとり,メチルオレンジを指示薬として再び水酸化ナトリウム液及び希硝酸で中和する(pH3)。水を加えて30mlとし,1-ブタノール-ベンゼン液10mlを加え約5分間激しく振り混ぜて静置し,水相は捨てる。有機相にピリジルアゾレソルシノール液10mlを正確に加え,約5分間激しく振り混ぜて静置し,水相を別の容器に移し約90分間以上放置したのち,空試験の液を対照液として545nmの吸光度を測定する。同時に標準バナジウム液を数段階に正確にとり,試料液の場合と同様に操作して作成した検量線からバナジウム(V)の量を求める。
5.23 ビウレット性窒素
5.23.1 硫酸銅法
A 適用範囲
尿素及び尿素を含有する肥料を対象とし,ビウレット性窒素をおおよそ0.05%以上含有するものに好適である。
B 装置
吸光光度分析装置
C 試薬液の調製
1)標準ビウレット液
ビウレット[(CO・NH2)2NH](110℃で恒量になるまで乾燥したもの)0.9812gを水に溶かして正確に100mlとする(この液1mlはビウレット性Nとして4mgを含有する)。
2)硫酸銅液
硫酸銅15gを水に溶かして1000mlとし,必要があればろ過する。
3)水酸化ナトリウム液
水酸化ナトリウム40gを水に溶かして1000mlとする。
4)硫酸アルミニウム液
硫酸アルミニウム[Al2(SO4)3・18H2O]2gを水に溶かして100mlとする。
D 試料液の調製
1)尿素
分析試料1~10g(ビウレット性Nとして20~60mgがよい)を100mlのメスフラスコに正確にとり,水約50mlを加えて溶かす。
2)尿素を含有する複合肥料
分析試料10~20g(ビウレット性Nとして20~60mgがよい)をビーカーに正確にとり,炭酸カルシウム1~2gを添加してよく混合し,75±1℃で4時間乾燥して水分及び遊離酸を除去したのち,ソックスレー抽出器を用いてアセトンでビウレットをことごとく浸出する。この浸出液を蒸発してアセトンを除去したのち,水を加えて溶かす。着色または混濁したときは脱色または除濁して清澄液とし,水で100mlのメスフラスコに移す。
(付記)
1.ビウレットの液の浸出は通常少なくとも20回以上の還流を必要とする。
2.脱色には活性炭または過酸化水素水を使用する。すなわち少量の活性炭(約0.05g)を加えてかき混ぜたのちろ過するか,30%過酸化水素水1~2mlを加えて蒸発乾固する。
3.除濁には硫酸アルミニウム液0.2~0.3mlを加え希水酸化ナトリウム液で中和してろ過する。
4.脂肪を多量に含有する原料を含有する試料では,上記のアセトン浸出液中に脂肪が溶出して以後の操作に支障を生ずるので,アセトンを蒸発して除去したのちジエチルエーテルを加えて脂肪を溶かし(ビウレットはジエチルエーテルに溶けない),デカンテーションによりこれを除去する必要がある。
5.硝酸アンモニウムを含有する試料では,アセトン浸出液を冷却して析出する固形物をろ別する。ろ紙上の固形物は更にアセトンで数回洗浄し,そのろ液の量を150ml以内にとどめる。このろ液を蒸発してアセトンを除去したのち,以下本文により試料液を作成する。なおこの場合の定量法は硫酸銅法に限り,銅錯塩法は適用できない。
6.発色を妨害する農薬を含有する試料にあっては,尿素の場合には水約50mlに溶かし,また尿素を含有する複合肥料の場合にはアセトンを蒸発させたのち水約50mlに溶かし,塩酸(1+2)3ml及びベンゼン20~30mlで抽出除去する(ただし水に溶けないときは単にろ別・水洗すればよい)。分離液は100mlのメスフラスコに受け,これに活性炭約0.05gを加えて脱色したのち標線まで水を加えて乾燥ろ紙でろ過し,このろ液50mlを100mlのメスフラスコに正確にとり,水酸化ナトリウム液で中和して試料液とする。
E 定量
試料液に水酸化ナトリウム液20mlを加え,更に硫酸銅液20mlを加えて発色させ,標線まで水を加えてよく振り混ぜ,約30分間放置後沈殿を遠心分離し,その上澄み液をとり波長540nm付近の吸光度を測定する。別に標準ビウレット液を数段階に正確にとり,試料液の場合と同一条件で操作して作成した検量線からビウレット性窒素(N)の量を求める。
(付記)
試料中にアンモニアまたは炭酸アンモニウムを多量に含有する場合には,分析試料をビーカーにとり,水数滴を加えて潤し水浴上で蒸発してあらかじめ揮発物を除去する必要がある。
5.23.2 銅錯塩法
A 適用範囲
5.23.1と同様であるが,本法は試料液中にアンモニア性窒素(N)が2mg以上存在する場合には適用できない。
B 装置
吸光光度分析装置
C 試薬液の調製
1)標準ビウレット液
5.23.1.C.1)による。
2)銅錯塩液
0.8%水酸化ナトリウム液約400mlに酒石酸カリウムナトリウム(KNaC4H4O6・4H2O)40gを溶かしたのち,硫酸銅10gを溶かし,更に0.8%水酸化ナトリウム液を加えて1000mlとする。
D 試料液の調製
5.23.1.Cによる。
E 定量
試料液に銅錯塩液40mlを加えて発色させ,標線まで水を加えてよく振り混ぜたのち,空試験の液を対照液として波長540nm付近の吸光度を測定する。別に標準ビウレット液を数段階に正確にとり,試料液の場合と同一条件で操作して作成した検量線からビウレット性窒素(N)の量を求める。
(付記)
本法において試料液の混濁はあらかじめ除去しなければならない。ただし発色試薬を加えることにより混濁を生ずる場合には本法は適用し難い。
5.23.3 原子吸光測光法
A 適用範囲
尿素及び尿素を含有する肥料を対象とする。ビウレットと結合した銅を5.18.2に準じて測定しビウレットを間接的に求める。
B 装置
原子吸光分析装置 銅中空陰極ランプ。
C 試薬液の調製
1)標準ビウレット液
5.23.1.C.1)による。
2)標準銅液
5.18.1.C.1)により作成した標準銅原液の一定量を使用に際して水で正確に希釈し,1ml中に銅として1~4μgの濃度で数段階に標準液を作成する。
3)硫酸銅液
5.23.1.C.2)による。
4)緩衝液(pH13.4)
水酸化カリウム24.6g及び塩化カリウム30gを水に溶かして1000mlとする。
5)デンプン液
溶性デンプン1gに冷水10mlを加え,かき混ぜてペースト状とする。この液を,シュウ酸1gを水150mlに溶かして沸騰させた中に徐々に加え,更に液が澄明になるまで煮沸する。冷却後水を加えて200mlとする。1週間は使用することができる。
6)指示薬
ブロムクレゾールパープル ブロムクレゾールパープル0.1gを0.1M水酸化ナトリウム液19mlに溶かし水を加えて250mlとする。
D 試料液の調製
1)尿素
分析試料1~5g(ビウレット性Nとして20mg以下)を100mlのメスフラスコに正確にとり,水約30mlを加えて溶かす。
2)尿素を含有する複合肥料
分析試料1~5g(ビウレット性Nとして20mg以下)をビーカーに正確にとり,分析試料1g当たり1mlの水を加えて加温する。アルコール65mlを加え,指示薬としてブロムクレゾールパープル7滴を加えて,20%水酸化カリウム液で青色が初めて現れるまで(pH6~7)中和する。熱板上で煮沸するまで加熱し,冷却する。もし青色が変わったときには再び中和する。ろ過器にろ紙パルプを敷いてあらかじめアルコールで洗浄してから試料液を減圧ろ過し,ろ液は100mlのメスフラスコに集める。(この際ろ液が澄明でない場合には,試料液に塩酸を加え,再びpHを6~7となるように調整する。)アルコールで洗浄し,更に標線までアルコールを加える。
E 定量
尿素の場合には試料液の全量,複合肥料の場合には試料液25mlを100mlのメスフラスコに正確にとり,アルコール25mlを加え,マグネチックスターラーで激しくかき混ぜながらデンプン液2ml,硫酸銅液10ml及び緩衝液20mlを加える。標線まで水を加え,よく振り混ぜたのち約10分間放置する。乾燥ガラスろ過器(17G3)などで約50mlを減圧ろ過し,そのろ液25ml正確にとって250mlのメスフラスコに移す。これに1M塩酸5ml及び水を標線まで加え,5.18.2.Eの「原子吸光分析装置により」以下により銅(Cu)の量を求める。なお,この際に用いる標準銅液には試料液と同量になるように,アルコール,デンプン液,緩衝液及び1M塩酸を加える必要がある。
同時に標準ビウレット液の一定量ずつ(ビウレット性Nとして2~5mg)を数段階に100mlのメスフラスコに正確にとり,水で液量を約30mlとしたのち,上記「アルコール25mlを加え」以下により銅(Cu)の量を求めビウレット性Nと銅の関係式を算出し,これより試料液中のビウレット性窒素(N)の量を求める。
5.24 ヒ素
5.24.1 ジエチルジチオカルバミン酸銀法
A 適用範囲
硫酸を原料として使用した肥料を主として対象とするが,その他の試料にも適用できる。
B 装置
1)水酸化ヒ素発生装置
JIS K 0102の61.1に示された装置に準ずるものであり,共通すり合わせのものがよい。
2)吸光光度分析装置
C 試薬液の調製
1)標準ヒ素液
標準試薬三酸化二ヒ素(As2O3)(105℃に3~4時間保ち硫酸デシケーター中で放冷したもの)1.320gをビーカーにとり,0.4%水酸化ナトリウム液約200mlを加え加熱して溶かし,冷却後フェノールフタレインを指示薬として希塩酸で中和し水を加えて正確に1000mlとし,標準ヒ素原液を作成する(この液1mlはAsとして1mgを含有する)。使用に際してこの原液の一定量を水で正確に1000倍に希釈する(この液1mlはAsとして1μgを含有する)。
2)ジエチルジチオカルバミン酸銀液
ジエチルジチオカルバミン酸銀[Ag(C2H5)2NCS2]0.5gをピリジン100mlに溶かし冷暗所に貯蔵する。または,ジエチルジチオカルバミン酸銀0.5gとブルシン(C23H26O4N2・2H2O)0.1gをクロロホルム200mlに溶かし(一夜間放置するとよい),冷暗所に貯蔵する。
3)塩化スズ(Ⅱ)液
塩化スズ(Ⅱ)(SnCl2・2H2O)15gを塩酸(1+1)100mlに溶かしたのち,少量の粒状スズを加えて着色瓶に貯蔵する。
4)ヨウ化カリウム液
ヨウ化カリウム(KI)200gを水に溶かして1000mlとする。
5)酢酸鉛ガラス綿
ガラス綿を10%酢酸鉛液で潤したのち風乾する。
6)指示薬
フェノールフタレイン 4.1.5.1.B.5).ⅲ)による。
D 試料液の調製
a ヒ素全量
1)無機質肥料
分析試料1~5gをビーカー(磁製がよい)に正確にとり,水で潤したのち硫酸2ml,硝酸約5ml及び過塩素酸20mlを加え,加熱して過塩素酸の白煙が発生し更に乾固近くになるまで濃縮する。放冷後水を加えて溶かし,冷却後100~200mlのメスフラスコに移し標線まで水を加えて乾燥ろ紙でろ過する。
2)有機物を含有する肥料
分析試料2gをビーカー(磁製がよい)に正確にとり,水で潤したのち硫酸2ml,硝酸5ml及び過塩素酸20mlを加え,時計皿で覆い砂浴上で加熱する。乾固近くになったならば,更に少量の硝酸及び過塩素酸を加え,加熱を続けて6時間分解したのち,前記1)の「加熱して過塩素酸の白煙が発生し」以下に従い操作する。
3)リン鉱石
分析試料1~5gを磁製ビーカーに正確にとり,水で潤したのち硝酸約5ml及び過塩素酸20mlを加え,次に前記1)の「加熱して過塩素酸の白煙が発生し」以下に従い操作する。
(付記)
硫酸塩類などの肥料の場合には,分析試料1gを三角フラスコに正確にとり,塩酸(1+1)20mlを加え環流冷却器を付けて15分間煮沸したのち放冷し,これを試料液としても差し支えない。
b 水溶性ヒ素
5.6.1.D.bによる。
E 定量
試料液の一定量(Asとして1~20μgがよく,液量は40ml以下)を80mlの水素化ヒ素発生瓶に正確にとり,水を加えて約40mlとし塩酸の量が10mlとなるように不足量を補い,ヨウ化カリウム液2mlを加え振り混ぜて数分間放置し,更に塩化スズ(Ⅱ)液1ml[鉄が多量に共存するときはアスコルビン酸1g及び塩化スズ(Ⅱ)液2mlを加える]を加え,振り混ぜて約10分間放置する。これに無ヒ素亜鉛(径1~1.5mm)2.5gを加え,発生するガスを酢酸鉛ガラス綿を軽く詰めたガラス管及びジエチルジチオカルバミン酸銀液5mlを正確に入れた吸収管に導くようにする。これらの発生瓶及びガラス管は気密に連結し,25℃前後で約45分間作用させる。吸収管内の発色液につき,同時に操作された空試験の液を対照液として波長510nm付近の吸光度を測定する。別に標準ヒ素液を数段階に正確にとり,試料液の場合と同時に同一条件で操作して作成した検量線からヒ素(As)の量を求める。
5.24.2 原子吸光測光法
A 適用範囲
ヒ素を水素化ヒ素としたのち,アルゴン-水素フレームで,または高温に加熱した吸収セルに導き原子吸光測光する。装置,条件によって差はあるが,試料液中で5~50ng/ml(アルゴン-水素フレーム法)または1~10ng/ml(加熱吸収セル法)のヒ素を測定できる。
B 装置
1)水素化ヒ素発生装置
JIS K 0102の61.2に示された亜鉛粉末を用いる装置,あるいはテトラヒドロホウ酸ナトリウムを用いる装置。
2)原子吸光分析装置
ヒ素中空陰極ランプ。アルゴン-水素フレーム,または加熱吸収セル付きのもの。
C 試薬液の調製
1)標準ヒ素液
5.24.1.C.1)による。
2)塩化スズ(Ⅱ)液
塩化スズ(Ⅱ)(SnCl2・2H2O)10gに塩酸100mlを加えて溶かす。
3)ヨウ化カリウム液
5.24.1.C.4)による。
4)テトラヒドロホウ酸ナトリウム
テトラヒドロホウ酸ナトリウム(NaBH4)10g及び水酸化ナトリウム5gを水に溶かして1000mlとする。使用の都度作成する。
D 試料液の調製
5.24.1.Dによる。
(付記)
下水汚泥などの有機物を含有する肥料であってヒ素全量を定量する場合には,5.24.1.D.a.2)によって分解したのち,塩酸(1+5)25mlを加え加熱して溶かし,冷却後100mlのメスフラスコに移して標線まで水を加える。
E 定量
1)アルゴン-水素フレーム法
試料液の一定量(Asとして0.1~1μgがよい)を水素化ヒ素発生装置の反応容器に正確にとり,水を加えて15mlとする。これに8M塩酸10ml,ヨウ化カリウム液1ml及び塩化スズ(Ⅱ)液1mlを加えて振り混ぜ約15分間放置する。水素化ヒ素発生装置と原子吸光分析装置を接続し,系内の空気をアルゴンで置換したのち,亜鉛粉末(ヒ素分析用)1.0gを手早く反応容器中に加え,マグネチックスターラーでかき混ぜ水素化ヒ素及び水素を発生させ,その一定量を捕集する。原子吸光分析装置と接続するコックを開き発生したガスをアルゴン-水素フレームに導き,波長193.7nmの吸光度を測定する。同時に標準ヒ素液を数段階に正確にとり,試料液の場合と同一条件で操作して作成した検量線からヒ素(As)の量を求める。
全操作にわたり空試験を必ず行い,結果を補正する。
(付記)
亜鉛粉末中のヒ素の量は無視できないので,一定量をあらかじめ成型するか,オブラートに包み一時に加え,亜鉛粉末についての空試験値を必ず補正する必要がある。
2)加熱吸収セル法
試料液(Asとして1~50ng/mlがよい),テトラヒドロホウ酸ナトリウム液及び塩酸(1+5)をそれぞれペリスタポンプで反応容器中に連続的に送り込み,発生する水素化ヒ素を含むガスを1000℃前後に加熱した吸収セルに導き,前記1)の「波長193.7nmの吸光度を」以下によりヒ素(As)の濃度を求める。
(付記)
2.5.14.2.Eに記載したビスムチオールⅡ-クロロホルム系でセレンと同時にヒ素も抽出できるので,このクロロホルム相の10~20μl(Asとして25~200ng)をフレームレス原子化装置に正確にとり,193.7nmの吸光度を測定してもよい。この場合の測定条件の一例を示すと,乾燥温度80℃,灰化温度1200℃で40秒,原子化温度2400℃である。
5.25 腐植酸(酸不溶-アルカリ可溶分)
5.25.1 塩酸-水酸化ナトリウム法
A 適用範囲
腐植酸塩肥料等の腐植成分の評価のため,酸・アルカリ処理により腐植酸を定量する簡易な方法である。腐植酸の本質は必ずしも明確なものではないが,ここにいう腐植酸とは,土壌の腐植物質の分画法の変法により定量される成分である。
B 装置
分析試料1gを200~300ml容の三角フラスコに正確にとり,塩酸(1+9)50mlを加え,1分間30~40回回転の振り混ぜ機で1時間振り混ぜたのち,1分間2500回回転以上で5分間以上遠心分離し,上澄み液をデカントする。次に遠心分離管内の沈殿物に水を加えてかき混ぜ,同様に遠心分離し,上澄み液を除く。さらにこの洗浄操作を3回繰り返す。あらかじめ重さを正確に量ったるつぼ型ガラスろ過器(1G4)へ不溶解物を水でことごとく洗い込んでろ過し,105~110℃で3時間乾燥したのち,重さを正確に量る(不溶解物:W1g)。
別に分析試料1gを同様に処理して得られた遠心分離管内の不溶解物を200~300ml容の三角フラスコに少量の水で洗い込み,1%水酸化ナトリウム液50mlを加え,1分間30~40回回転の振り混ぜ機で1時間振り混ぜたのち,上記と同様に遠心分離・水洗処理を行う。不溶解物を乾燥後平形量り瓶に移し,105~110℃で3時間乾燥したのち,重さを正確に量る(不溶解物:W2g)。次式により腐植酸(酸不溶-アルカリ可溶分)の量を算出する。
腐植酸(酸不溶-アルカリ可溶分)(%)=(W1-W2)×100
5.26 フッ素
5.26.1 アリザリンコンプレクソン法
A 適用範囲
リン鉱石を主として対象とするが,その他フッ素を含有する試料に広く適用できる。
B 装置
1)フッ素蒸留装置
JIS K 0102の34.1に示されたものに準ずる。
2)吸光光度分析装置
C 試薬液の調製
1)標準フッ素液
標準試薬フッ化ナトリウム(NaF)(白金るつぼ中で500~550℃に40~50分間保ち硫酸デシケーター中で放冷したもの)2.210gを水に溶かして正確に1000mlとし,この原液の一定量を水で正確に100倍に希釈しポリエチレン瓶に貯蔵する(この液1mlはFとして10μgを含有する)。
2)ランタン-アリザリンコンプレクソン液
使用に際して混成試薬アルフッソンの10%水溶液を作成する。
3)水酸化ナトリウム液
水酸化ナトリウム10gを水に溶かして1000mlとする。
4)指示薬
メチルレッド メチルレッド0.2gを90%アルコールに溶かして1000mlとしろ過する。
D 試料液の調製
分析試料0.5~1g(Fとして1~25mgがよい)を分解フラスコに正確にとり,少量のケイ砂末及び硫酸銀(塩素を沈殿させるのに足りる量)を加え,水蒸気導入ガラス管及び温度計を備え付けたゴム栓をし,そのガラス管及び温度計の下端は液中に浸るようにする。過塩素酸約10mlを加えて密栓し,別に沸騰するフラスコより水蒸気を分解フラスコに導入しフッ素をケイフッ化水素酸(H2SiF6)として留出させ,冷却器を通して500mlのメスフラスコに受ける。分解フラスコは小炎で熱して徐々に温度を上げ,液温140~145℃で留出液が約200mlになるまで蒸留したのち,メチルレッドを指示薬として水酸化ナトリウム液で中和し,標線まで水を加える。
(付記)
1.試料に有機物または硫化物を多量に含有する場合には,分析試料の一定量を磁製るつぼに正確にとり,有機物が多いときはフッ素を含有しない石灰乳を加えて乾燥し低熱で灰化し,また硫化物が多いときは低熱で硫黄分を駆遂したのち,分解フラスコに移す。有機物(0.2g以下)または硫化物が少量の場合には,分析試料をそのまま分解フラスコにとり,蒸留に先だって少量の過マンガン酸カリウム飽和液を加えて酸化すればよい。
2.試料にアルミニウムを多量(Alとして25mg以上)に含有する場合には,分析試料0.5~1gをめのう乳鉢に正確にとってよく粉砕し,同量のよく粉砕したケイ砂末(ケイ素を含有する試料では少量でよい)を加え,以下4.4.1.B.a.2)によりケイ素,アルミニウムなどを除去したろ液を集めたフッ素浸出液を蒸発濃縮したのち,分解フラスコに移す。
E 定量
試料液の一定量(Fとして1~50μg,液量10ml以下)を25mlのメスフラスコに正確にとり,水を加えて約10mlとし,これにランタン-アリザリンコンプレクソン液3mlを正確に加え,更にアセトン10mlを加え,冷却後標線まで水を加えて振り混ぜる。約1.5時間放置後再び振り混ぜ,直ちに空試験の液を対照液として波長620nm付近の吸光度を測定する。別に標準フッ素液を数段階に正確にとり,試料液の場合と同時に同一条件で操作して作成した検量線からフッ素(F)の量を求める。
5.26.2 イオン選択性電極法
A 適用範囲
フッ素を含有する試料に適用する。
B 装置
イオン計 フッ素イオン活量選択性電極を付けたもの。
C 試薬液の調製
1)標準フッ素液
5.26.1.C.1)により作成した標準フッ素原液を用いる(この液1mlはFとして1mgを含有する)。
2)クエン酸塩液
クエン酸リチウム(Li3C6H5O7・4H2O)420g[またはクエン酸ナトリウム(Na3C6H5O7・2H2O)440g]を適量の水に加温して溶かし,塩酸(1+1)を加えてpH5.0~5.5に調整し水で1000mlとする。
3)水酸化ナトリウム
5.26.1.C.3)による。
4)指示薬
メチルレッド 5.26.1.C.4)による。
D 試料液の調製
5.26.1.Dによる。
(付記)
試料によっては次の方法によることができる。
分析試料をめのう乳鉢により,よく粉砕したのち,その0.3gを250mlのメスフラスコに正確にとり,塩酸(1+1)5ml及び水約150mlを加え,1分間30~40回回転の振り混ぜ機で1時間振り混ぜたのち,メチルレッドを指示薬として水酸化ナトリウム液で中和し,標線まで水を加える。
E 定量
試料液25mlを100mlのメスフラスコに正確にとり,クエン酸塩液10mlを加え標線まで水を加え,その50mlを小型ビーカーに正確にとり,クエン酸塩液50mlを加えイオン計の電位(mV)を読む。同時に標準フッ素液を一定濃度に希釈した液(F0.01~1000μg/ml)につき試料液の場合と同様に操作して作成した検量線(片対数方眼紙を用いフッ素濃度を対数軸にとる)からフッ素(F)の量を求める。共存成分の影響がある場合には,測定後の試料液10mlを100mlのメスフラスコに正確にとり,クエン酸塩液50mlを加え標線まで水を加え,同様にして電位を測定しフッ素(F)の量を求め,この操作を繰り返して希釈前後の測定値が同一になったならば,その値をフッ素(F)の量とする。
(付記)
5.26.1.Dによりフッ素を蒸留した場合には,クエン酸塩液の添加量を減らし,あるいは省略することができる。
5.27 モリブデン
5.27.1 チオシアン酸ナトリウム法
A 適用範囲
主として微量要素としてモリブデンを添加した肥料を対象とする。
B 装置
吸光光度分析装置
C 試薬液の調製
1)標準モリブデン液
特級三酸化モリブデン(MoO3)(硫酸デシケーター中で24時間以上乾燥したもの)1.500gを少量の水で潤し,水酸化ナトリウム約5gを加えて溶かしたのち,水を加えて正確に1000mlとし標準モリブデン原液を作成し(この液1mlはMoとして1mgを含有する),着色瓶に貯蔵する。使用に際してこの原液の一定量を水で正確に100倍に希釈する(この液1mlはMoとして10μgを含有する)。
2)チオシアン酸ナトリウム液
チオシアン酸ナトリウム(NaSCN)10gを水に溶かして100mlとする。
3)硫酸鉄(Ⅲ)液
硫酸鉄(Ⅲ)[Fe2(SO4)3・9H2O]5gを水及び硫酸(1+1)約10mlに溶かして100mlとする。
4)塩化スズ(Ⅱ)
塩化スズ(Ⅱ)(SnCl2・2H2O)100gを塩酸(1+1)400mlに加温して溶かしたのち,水を加えて1000mlとし,少量の粒状スズを加えて着色瓶に貯蔵する。
D 試料液の調製
a モリブデン全量
5.1.1.D.aによる。
b 水溶性モリブデン
5.1.1.D.bによる。
(付記)
作成した試料液に妨害となる有機物が存在するときは,あらかじめろ液の一定量を正確にとり,少量の硫酸及び硝酸で分解したのち試料液とする。
E 定量
試料液の一定量(Moとして10~300μgがよい)を100mlのメスフラスコに正確にとり,硫酸(1+1)5ml(硫酸で分解した試料液では含有する硫酸量だけ減ずる),過塩素酸5ml及び硫酸鉄(Ⅲ)液2mlを加えて混合し,更にチオシアン酸ナトリウム液16ml及び塩化スズ(Ⅱ)液10mlを試料液を振り混ぜながら徐々に順次加えたのち,標線まで水を加える。鉄による赤色が消失したのち,溶液が混濁する場合には[混濁がチオシアン酸銅(Ⅰ)によるときは1時間放置後に]遠心器により清澄にし,空試験の液を対照液として波長460nm付近の吸光度を測定する。別に標準モリブデン液を数段階に正確にとり,試料液の場合と同一条件で操作して作成した検量線からモリブデン(Mo)の量を求める。
(付記)
発色液の吸光度が0.1以下のときは,次に記載する酢酸=1-ブチルによる抽出を行う。
発色液の一部または全部(酢酸=1-ブチル10mlに対しMoとして1~15μgを含有する量)を小型分液漏斗に正確にとり,酢酸=1-ブチルの一定量(20または10ml)を正確に加えて1~2分間激しく振り混ぜ,静置後水相を捨て再び少量の水を加え,更に塩化スズ(Ⅱ)液を鉄による赤色が消失する程度(約10ml)を加えたのち軽く振り混ぜる。静置後水相を捨て,残留する酢酸=1-ブチル相につき(混濁するときは遠心器により清澄にし),本文の「空試験の液を対照液として」以下によりモリブデンを定量する。
5.27.2 原子吸光測光法
A 適用範囲
5.27.1と同様である。モリブデンの原子吸光測光法の感度は通常あまり高くないので,溶媒抽出と組み合わせることが必要である。
B 装置
原子吸光分析装置
C 試薬液の調製
1)標準モリブデン液
5.27.1.C.1)による。
2)8-キノリノール液
8-キノリノール(オキシン;C9H6N・OH)20gをビーカーにとり,酢酸50mlを加え水浴上で加熱して溶かしたのち,水を加えて100mlとする。
D 試料液の調製
5.27.1.Dによる。
E 定量
試料液の一定量(Moとして10~200μgがよい)を100mlのメスフラスコに正確にとり,1M塩酸10ml,8-キノリノール液5ml及び水を加えて約80mlとし,よく振り混ぜたのち少時放置する。2-ヘプタノンまたは4-メチル-2-ペンタノン(イソブチルメチルケトン)5~10mlを正確に加え,1~2分間激しく振り混ぜたのち静置する。これに水約20mlを加え,上層の有機溶媒相につき,原子吸光分析装置により波長313.3nmの吸光度を測定する。同時に標準モリブデン液を数段階に正確にとり,試料液の場合と同一条件で操作して作成した検量線からモリブデン(Mo)の量を求める。
5.28 ウラン
5.28.1 8-キノリノール法
A 適用範囲
リン鉱石を対象とする。ウラン含量0.005~0.06%のものに適用する。
B 装置
吸光光度分析装置
C 試薬液の調製
1)標準ウラン液
金属ウラン[硝酸(1+2),水,アルコール,ジエチルエーテルで順次洗浄し乾燥したもの]1gをビーカーに正確にとり,硝酸(1+1)20mlを加え加熱して溶かし,1000mlのメスフラスコに移し標線まで水を加える。この原液1mlはウラン1mgを含有する。この原液10mlを1000mlのメスフラスコに正確にとり,硝酸(1+1)14mlを加え,更に標線まで水を加える。この液1mlはウラン10μgを含有する。
2)8-キノリノール液
8-キノリノール(オキシン;C9H6N・OH)10gをクロロホルム1000mlに溶かし,着色瓶に貯蔵する。
3)リン酸トリ-n-ブチル-ケロシン液
リン酸トリ-n-ブチル3部とケロシン7部を混合し,1%水酸化ナトリウム,水,次いで硝酸(1+2)でそれぞれ2回ずつ振り混ぜ,硝酸で飽和させる。
4)硝酸-硝酸ナトリウム洗浄液
硝酸ナトリウム500gを温水に溶かし,硝酸70ml及び水に加えて1000mlとする。
5)硫酸アンモニウム液
硫酸アンモニウム250gを水に溶かして1000mlとする。
6)エチレンジアミン四酢酸二ナトリウム液
エチレンジアミン四酢酸二ナトリウム(Na2H2C10H12N2O8・2H2O)50gを水に溶かして1000mlとする。
7)水酸化ナトリウム液
水酸化ナトリウム300gを水に溶かして1000mlとする。
D 試料液の調製
リン鉱石
分析試料0.5~1gをトールビーカーに正確にとり,少量の水で潤す。硝酸30mlを加え加熱して蒸発し,乾固に至らせる。(分解が不完全なときは硝酸20mlを加え再び乾固する。)放冷後,硝酸(1+4)5mlを加え,沸騰し始めるまで加熱し,ろ過し温水で洗浄してろ液を小型ビーカーに集める。このろ液を蒸発し液量を約5mlとして放冷する。
E 定量
試料液全量を水酸化ナトリウム液で沈殿がわずかにできるまで中和し,硝酸(1+1)を滴下して生成した沈殿を溶かし,更にその4mlを過剰に加える。この液を小型分液漏斗に水で洗い込み,液量を約30mlとする。硝酸アルミニウム[Al(NO3)3・9H2O]20gを加え振り混ぜて溶かす。リン酸トリ-n-ブチル-ケロシン液25mlを加え,約2分間激しく振り混ぜウランを抽出する。静置したのち水相を捨て,有機相に硝酸-硝酸ナトリウム洗浄液20mlを加え,約2分間激しく振り混ぜて静置したのち,水相を捨て,再び同洗浄液10mlを加え同様に操作して洗浄する。次いで有機相に硫酸アンモニウム液25mlを加え,約2分間激しく振り混ぜてウランを逆抽出する。静置後水相を小型ビーカーに移し,有機相に硫酸アンモニウム液10mlを加え再び逆抽出して水相を合わせる。
この逆抽出した水溶液にアンモニア水(1+1)10ml及び水を加えて約60mlとし,穏やかに加熱し約25mlになるまで濃縮する。放冷後,エチレンジアミン四酢酸二ナトリウム液5mlを加え,ガラス電極pH計を使いアンモニア水(1+50)または塩酸(1+50)を加えてpHを7.0~7.5とする。分液漏斗(200ml容)に水で移して液量を約60mlとする。8-キノリノール液20mlを正確に加え,約2分間激しく振り混ぜてウラン錯塩を抽出する。静置したのち有機相を脱脂綿でろ過し,そのろ液につき空試験の液を対照液とし波長380nmまたはその付近の吸光度を測定する。別に標準ウラン液を数段階に正確にとり,同様に操作して作成した検量線からウラン(U)の量を求める。
5.29 遊離硫酸(酸分)及び硫酸塩
5.29.1 遊離硫酸(酸分)
5.29.1.1 アルカリ法
A 適用範囲
硫酸塩類中の遊離硫酸の定量に適用するが,同時に硫酸一水素塩が共存する場合には滴定値が過大になることがある。
B 試薬液の調製
1)標準水酸化ナトリウム液
4.1.1.1.B.1)により0.1M水酸化ナトリウム溶液を作成する。
2)指示薬
メチルレッド 4.1.1.1.B.4).ⅱ)による。
C 試料液の調製
分析試料12.5gを250mlのメスフラスコに正確にとり,水約200mlを加え1分間30~40回回転の振り混ぜ機で30分間振り混ぜたのち,標線まで水を加えて直ちに乾燥ろ紙でろ過する。
D 定量
試料液100mlを三角フラスコに正確にとり,指示薬としてメチルレッド2~3滴を加えて標準水酸化ナトリウム液で滴定し,その滴定値よりこれに相当する硫酸(H2SO4)の量を算出して遊離硫酸(酸分)の量とする。
0.1M水酸化ナトリウム液1ml=4.904mgH2SO4
5.29.2 硫酸塩
5.29.2.1 塩化バリウム法
A 適用範囲
腐植酸塩等を主体とする肥料に硫酸塩の規制がある。硫酸塩中の硫酸の定量に適用する。
B 試薬液の調製
1)塩化バリウム法
4.1.1.1.B.3)による。
2)指示薬
メチルレッド 4.1.1.1.B.4).ⅱ)による。
C 試料液の調製
1) 腐植酸塩肥料
分析試料10gを250mlのメスフラスコに正確にとり,塩酸(1+9)150mlを加え1分間30~40回回転の振り混ぜ機で1時間振り混ぜたのち,標線まで水を加えて混合し,乾燥ろ紙でろ過する。
2)リグニン苦土(マグネシウム)肥料
分析試料5gを250mlのメスフラスコに正確にとり,水200mlを加え1分間30~40回回転の振り混ぜ機で30分間振り混ぜたのち,標線まで水を加えて乾燥ろ紙でろ過する。
3)セッコウ
分析試料1gを300ml容のトールビーカーに正確にとり,塩酸約30ml及び硝酸約10mlを加えて時計皿で覆い30分間煮沸し,冷却後250mlのメスフラスコに移し標線まで水を加えて乾燥ろ紙でろ過する。
D 定量
試料液50~100ml(SO3として10~200mgがよい)を500ml容のトールビーカーに正確にとり,次に4.1.1.1.B.2)の「水で約300mlに希釈し」以下により硫酸バリウムの重さを正確に量る。この量から以下により硫酸塩の量を計算する。
1)腐植酸アンモニウム肥料
係数0.5662を掛けて硫酸アンモニウム[(NH4)2SO4]の量を算出し,硫酸塩の量とする。
2)腐植酸カリウム肥料
係数0.7466を掛けて硫酸カリウム(K2SO4)の量を算出し,硫酸塩の量とする。
3)リグニン苦土(マグネシウム)肥料
係数0.4116を掛けて水溶性硫酸イオン(SO42-)の量を算出する。一方,この試料液については4.5.1によりカルシウム(または酸化カルシウム)の量を求め,次式により硫酸塩に由来するマグネシウム(または酸化マグネシウム)の量を算出する。
硫酸塩由来のマグネシウム(Mg)(%)=[水溶性硫酸イオン(%)-水溶性カルシウム(%)×96.064/40.078]×24.305/96.064
硫酸塩由来の酸化マグネシウム(MgO)(%)=[水溶性硫酸イオン(%)-水溶性酸化カルシウム(%)×96.064/56.077]×40.304/96.064
4)セッコウ
係数0.5833を掛けて硫酸カルシウム(セッコウ,CaSO4)の量を算出する。一方,この試料液について4.5.1によりカルシウム(または酸化カルシウム)の量を求め,これより硫酸カルシウムの量を算出する。
硫酸カルシウム(CaSO4)=カルシウム(Ca)×3.3969=酸化カルシウム(CaO)×2.4277
この両者の試算値のうち,小さいほうの値を硫酸カルシウム(CaSO4)の量とする。
5.30 遊離リン酸
5.30.1 アセトン-ジエチルエーテル法
A 適用範囲
過リン酸石灰及び(三)重過リン酸石灰を対象とする。
B 試薬液の調製
1)標準水酸化ナトリウム液
4.1.1.1.B.1)により0.1M水酸化ナトリウム溶液を作成する。
2)アセトン-ジエチルエーテル液
アセトン500mlとジエチルエーテル500mlを混合して共栓瓶に入れ,冷所に貯蔵する。
3)指示薬
ⅰ)メチルイエロー メチルイエロー0.1gを90%アルコールに溶かして100mlとする。
ⅱ)フェノールフタレイン 4.1.5.1.B.5).ⅲ)による。
C 試料液の調製
分析試料2gをフラスコに正確にとり,アセトン-ジエチルエーテル液100mlを正確に加え1分間30~40回回転の振り混ぜ機で30分間振り混ぜたのち,乾燥ろ紙で速やかにろ過する。
D 定量
試料液50mlをビーカーに正確にとり,70~80℃の水浴上でほとんど蒸発乾固したのち,温水約50mlを加えてよくかき混ぜる。次に指示薬としてメチルイエロー1滴を加え標準水酸化ナトリウム液で黄変するまで滴定し,更に指示薬としてフェノールフタレイン数滴を加えて滴定を続け,微桃色の発現を終点とし,その全滴定値よりリン酸(P2O5)の量を算出して遊離リン酸の量とする。
0.1M水酸化ナトリウム液1ml=3.549mgP2O5
(付記)
指示薬メチルイエローの添加は省略してもよい。
5.31 陽イオン交換容量(塩基置換容量)
5.31.1 酢酸アンモニウム法
A 適用範囲
岩石,粘度等を含む鉱物質資材に適する。
B 装置
図1に示すように,100ml容で10mlごとに目盛りのついた洗浄液容器(A),長さ4cm,内径0.3cmの脚を持った長さ12cm,内径1.3cmの浸透管(B)及び受器(C)を上中下に連結し,外気を遮断して流下式に溶液が浸透するようにしたものである。
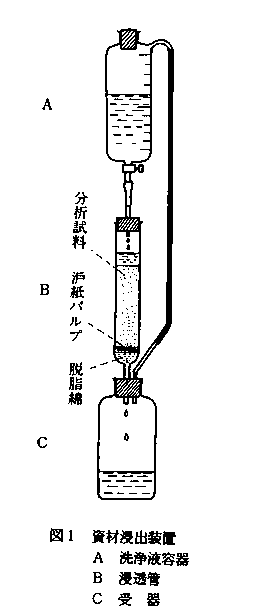
C 試薬液の調製
1)標準水酸化ナトリウム液
4.1.1.1.B.1)により0.1M溶液を作成する。
2)標準塩酸液
4.5.2.1.B.2)に準じて0.1M塩酸溶液を作成し,標準水酸化ナトリウム液の一定量を滴定してその濃度を標定する。
3)酢酸アンモニウム液
特級アンモニア水67mlに水を加えて正確に500mlとし,2Mアンモニア液を作成する。次に特級酢酸58mlに水を加えて正確に500mlとし,2M酢酸液を作成する。両液を容量比1対1の割合で混合したのち,ガラス電極法によりpH7.0となるようにアンモニア水または酢酸を添加して1M中性液を作成する。
4)アルコール液
80%アルコールを作成し,ブロムチモールブルーを指示薬としてpH7.0の色調(青緑色)になるまでアンモニア水で中和する。
5)指示薬
ⅰ)ブロムチモールブルー 4.1.1.1.B.4).ⅰ)による。
ⅱ)フェノールフタレイン 4.1.5.1.B.5).ⅲ)による。
D 定量
まず浸透管の下部に脱脂綿の小片を支持層として入れ,その上にろ紙を細かく切って熱水中でかき混ぜて作ったろ紙パルプを約5mmの厚さに詰めて平らなろ過面を作る。次に浸透管の下端に栓をして酢酸アンモニウム液数mlを入れ,そこに分析試料2~4gを正確に量って少量ずつ落下沈降させて充てんしたのち,栓を外して装置を組み立て,酢酸アンモニウム液で洗浄を始める。酢酸アンモニウム液は100mlを用い,4~20時間に浸透し終わるように滴下速度を調節する。浸透終了後少量のアルコール液で浸透管の内側上部を洗い落とし,更にアルコール液50mlで試料層を洗浄して過剰の酢酸アンモニウム液を除去する。このようにして得たNH4+で飽和された分析試料を10%塩化ナトリウム液100mlで洗浄してNH4+を交換浸出する。この浸出液中のアンモニア性窒素を4.1.2.1.Dにより水蒸気蒸留及びアルカリ滴定して定量し,乾物100g当たりのミリグラム当量として示し陽イオン交換容量(塩基置換容量)とする。
0.1M水酸化ナトリウム液1ml=0.1ミリグラム当量
(付記)
1.酢酸アンモニウム液の濃度とpHとについては正確に所要の値に調製する必要がある。一般に濃度やpHが高いほど得られる測定値は高くなる。ここで1M,pH7.0という条件を採用している。
2.蒸留用アルカリ剤には浸出された有機物の分解を考慮して弱アルカリ性の塩基性炭酸マグネシウム[4MgCO3・Mg(OH)2・nH2O]を用いる。
5.31.2 酢酸バリウム法
A 適用範囲
たい肥等の有機肥料に適する。
B 装置
図2に示すように,ウィットろ過器(内径12cm,高さ18cmのもの)の上部にシリコンゴム管を先端に付けた漏斗型ガラスろ過器を取り付ける。
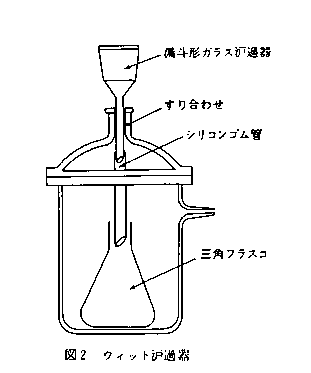
C 試薬液の調製
1)標準水酸化ナトリウム液
4.1.1.1.B.1)に準じて0.05M溶液を作成し,その濃度を標定して補正係数(F)を算出する。
2)酢酸バリウム液
酢酸バリウム[Ba(CH3CO2)2]127.71gを水900mlに溶かし,希酢酸または水酸化バリウム液を加えてガラス電極でpH7.0に調整したのち,水を加えて正確の1000mlとし,必要があればろ過する。
3)指示薬
チモールブルー 4.1.2.3.B.6).ⅲ)による。
D 定量
風乾後微粉砕した分析試料0.2g程度(乾物としてWg)を漏斗型ガラスろ過器に正確にとり,ウィットろ過器に取り付け,先端をピンチコックで閉じておく。0.05M塩酸25mlを加え,ガラス棒で時々かき混ぜながら20分間放置する。ウィットろ過器に300ml用の三角フラスコを入れ,ピンチコックを外し,緩やかに吸引しながらろ過する。この操作を更に1回繰り返す(ただし放置時間は約10分間でよい)。ガラスろ過器上の分析試料を吸引しながら塩素イオンの反応がなくなるまで水で洗浄する(通常150ml程度の洗浄で十分である)。
次にウィットろ過器内に別の300ml容の三角フラスコを入れ,ピンチコックを閉じてから酢酸バリウム液25mlを正確に加え,ガラス棒で時々かき混ぜながら1時間放置した後,ピンチコックを外し,吸引しながら三角フラスコにろ過する。この操作を更に1回繰り返したのち(ただし放置時間は約10分間でよい),水で十分洗浄する(通常150ml程度の洗浄で十分である)。これらのろ液をチモールブルーを指示薬として標準水酸化ナトリウム液で滴定する(t1ml)。空試験として酢酸バリウム液50mlに水150mlを加えたものについても,同様にして標準水酸化ナトリウム液で滴定する(t0ml)。次式により乾物100g当たりの陽イオン交換容量(ミリグラム当量)を算出する。
陽イオン交換容量(meq/乾物100g)=(t1-t0)×5F/W
5.32 溶出試験(重金属等の)
5.32.1 水溶出法
A 適用範囲
金属等を含む産業廃棄物に係る判定基準を定める総理府令(昭和48年2月総理府令第5号)の基準の検定方法(昭和48年2月環境庁告示第13号)で,じんかい灰,汚泥肥料,たい肥,石灰処理肥料等の特殊肥料に適用する。試験の内容は,水で溶出された1)アルキル水銀化合物,2)水銀またはその化合物,3)カドミウムまたはその化合物,4)鉛またはその化合物,5)有機リン化合物,6)6価クロム化合物,7)ヒ素またはその化合物,8)シアン化合物,9)ポリ塩化ビフェニル(PCB),10)トリクロロエチレン及び11)テトロクロロエチレンの11物質を検定するものである。
B 試料液の調製
a 前記Aの1)~9)の物質
分析試料(風乾しない現物)の一定量(g)と塩酸を加えてpH5.8~6.3とした水(ml)とを重量体積比1:10の割合で混合し,かつその混合液が500ml以上となるようにし,常温(約20℃)・常圧(約1気圧)において水平振り混ぜ機(振り混ぜ回数1分間約200回,振り混ぜ幅4~5cm)で6時間連続して振り混ぜたのち,孔径1μmのガラス繊維ろ紙でろ過して(ろ過が著しく困難な場合には,1分間約3000回回転で20分間遠心分離したのちの上澄み液を)試料液とする。
b 前記Aの10)及び11)の物質
分析試料(風乾しない現物)の一定量(g)と水酸化ナトリウム液または塩酸を加えてpH5.8~6.3とした水(ml)とを重量体積比1:10の割合に,かつその混合液が500ml以上となるように,混合液に対するヘッドスペースの少ないねじ口付き三角フラスコにとり,直ちに密栓し,常温(約20℃)に保ちながらマグネチックスターラーで4時間連続して振り混ぜる。10~30分間静置後,上澄み液約20mlを,あらかじめろ紙を装着したろ紙ホルダーを接続しておいた20ml容のガラス製注射筒の外筒に静かにとり,注射筒の内筒を押し,空気及び初めの数mlを排出し,次にろ液10mlを25ml容の共栓付き試験管に正確にとって試料液とする。
C 定量
1)アルキル水銀化合物
試料液の一定量を正確にとり,「水質汚濁に係る環境基準について」(昭和46年12月環境庁告示第59号(以下「告示」という))付表4の第1 ガスクロマトグラフィー及び第2 薄層クロマトグラフ分離-原子吸光測光法によりアルキル水銀(Hg)の濃度(mg/1)を求める。定量限界はアルキルHgとして0.0005mg/1である。
2)水銀またはその化合物
試料液の一定量を正確にとり,「告示」付表3または5.12.1の還元気化原子吸光測光法により,水銀(Hg)の濃度(mg/l)を求める。定量限界はHgとして0.0005mg/lである。
3)カドミウムまたはその化合物
試料液の一定量を正確にとり,JIS K 0102 55または5.6.1の原子吸光測光法によりカドミウム(Cd)の濃度(mg/l)を求める。定量限界はCdとして0.002mg/lである。
4)鉛またはその化合物
試料液の一定量を正確にとり,JIS K 0102 54または5.19.1の原子吸光測光法により鉛(Pb)の濃度(mg/l)を求める。定量限界はPbとして0.05mg/lである。
5)有機リン酸化合物
試料液の一定量を正確にとり,「告示」付表1のガラスクロマトグラフィー等により(ただしメチルジメトンにあっては,「告示」付表2のモリブデン酸青吸光光度法により)有機リン化合物(パラチオン,メチルパラチオン,メチルジメトンなど)の濃度(mg/l)を求める。定量限界は有機リン化合物として0.1mg/lである。
6)6価クロム化合物
試料液の一定量を正確にとり,JIS K 0102 65.2または5.8.2の原子吸光測光法により6価クロム[Cr(VI)]の濃度(mg/l)を求める。定量限界はCr(VI)として0.05mg/lである。
7)ヒ素またはその化合物
試料液の一定量を正確にとり,JIS K 0102 61または5.24.1のジエチルジチオカルバミン酸銀吸光光度法または5.24.2の原子吸光測光法によりヒ素(As)の濃度(mg/l)を求める。定量限界はAsとして0.01g/lである。
8)シアン化合物
試料液の一定量を正確にとり,JIS K 0102 38(ただし38.1.1を除く)ピラゾロン吸光光度法またはイオン電極法によりシアン(CN)の濃度(mg/l)を求める。定量限界はCNとして0.01mg/lである。
9)ポリ塩化ビフェニル(PCB)
試料液の一定量を正確にとり,「告示」付表5のガスクロマトグラフィーによりポリ塩化ビフェニル(PCB)の濃度を求める。定量限界はPCBとして0.0005mg/lである。
10)トリクロロエチレン
試料液につき,前記Aの昭和48年2月環境庁告示第13号別表第三のガスクロマトグラフィーによりトリクロロエチレンの濃度(mg/l)を求める。定量限界はトリクロロエチレンとして0.05mg/lである。
11)テトラクロロエチレン
前項10)によりテトラクロロエチレンの濃度(mg/l)を求める。定量限界はテトラクロロエチレンとして0.05mg/lである。
5.33 溶出率(被覆肥料の)
5.33.1 水中静置法
A 適用範囲
被覆肥料に適用する。
B 装置
恒温器 30±1℃に調製する。
C 試薬液の調製
1)窒素用試薬液
4.1の各項による。
2)リン(リン酸)用試薬液
4.2の各項による。
3)カリウム(加里)用試薬液
4.3の各項による。
D 試料液の調製
a 初期溶出率
試料(有姿のもの)12.5gを300ml容の三角フラスコに正確にとり,30℃の水250mlを正確に加えて密栓し,恒温器中で24時間静置したのち乾燥ろ紙でろ過する。
b 微分溶出率
前記aと同様に操作し恒温器中で一定期間(例えば7日間)静置したのち乾燥ろ紙でろ過する。
E 定量
a 初期溶出率
別に2.2によって調製した分析試料について,前記と同様に各成分をそれぞれ定量して試料中の各含有成分量を求め,それらの値で各初期溶出成分量を割って初期溶出率(%)を算出する。
b 微分溶出率
試料液の一定量を正確にとり,前記aの場合と同様に窒素全量,アンモニア性窒素,硝酸性窒素,水溶性リン(リン酸),水溶性カリウム(加里)をそれぞれ定量し,各積算溶出成分量を求める。この値より各初期溶出成分量を差し引き,試料中の各含有成分量及び静置日数より1を引いた値でそれぞれ割って微分溶出率(%)を算出する。
6 硝酸化成抑制材
6.1 2-アミノ-4-クロロ-6-メチルピリミジン(AM)
6.1.1 紫外部吸光光度法
A 適用範囲
2-アミノ-4-クロロ-6-メチルピリミジン(以下AMと略す)を含有する肥料に適用する。硝酸塩または有機物を含有する肥料には適用できない場合がある。
B 装置
吸光光度分析装置 紫外部における測定ができる装置であり,吸収セルは石英製とする。
C 試薬液の調製
1)標準AM液
AM(C5H6N3Cl)0.1gを正確にとり,水に溶かして正確に1000mlとする(この液1mlはC5H6N3Clとして0.1mgを含有する)。
2)塩化カリウム緩衝液
塩化カリウム93.2gを水500mlに溶かし,塩酸215ml及び水を加えて1000mlとする。
D 試料液の調製
分析試料5gを小型乳鉢に正確にとり,少量の水を加えてよくすりつぶし,その上澄み液を500mlのメスフラスコに移し,更にこの操作を3回反復したのち,乳鉢内の不溶解物を水でことごとくメスフラスコに移し,水を加えて約400mlとし,1分間30~40回回転の振り混ぜ機で30分間振り混ぜたのち,標線まで水を加えて乾燥ろ紙でろ過する。
E 定量
試料液の一定量(C5H6N3Clとして0.5~2.0mg相当量)を100mlのメスフラスコに正確にとり,塩化カリウム緩衝液20mlを加え,標線まで水を加え,塩化カリウム緩衝液を水で5倍に希釈した空試験の液を対照液として波長302,292及び312nmの吸光度をそれぞれ測定し,各測定値をa,b及びcとし,補正吸光度2a-(b+c)を算出する。別に標準AM液を数段階(C5H6N3Clとして0.5~2.0mg相当量)に正確にとり,試料液の場合と同一条件で操作して算出した補正吸光度より作成した検量線からAM(C5H6N3Cl)の量を求める。
6.1.2 高速液体クロマトグラフィー
A 適用範囲
2-アミノ-4-クロロ-6-メチルピリミジン(以下AMと略す)を含有する肥料に適用する。
B 装置
紫外部吸光光度分析装置付き高速液体クロマトグラフ
C 試薬液の調製
1)標準AM液
6.1.1.C.1)により作成した液を標準AM原液とする。使用に際してこの原液の一定量を抽出溶媒で正確に20~5倍に希釈する(この液1mlはC5H6N3Clとして5~20μgを含有する)。
2)抽出溶媒
メチルアルコール1容と水1容を混合する。
D 試料液の調製
分析試料1g(C5H6N3Clとして0.5mg相当量以上)を200ml容の共栓付き三角フラスコに正確にとり,抽出溶媒100mlを正確に加え,マグネチックスターラーで30分間かき混ぜたのち,1分間3000回回転で5分間遠心分離する。上澄み液の一定量を抽出溶媒で正確に一定倍に希釈し,孔径0.5μm以下のメンブランフィルターでろ過する。
(付記)
有機物を含有する肥料の場合には,上記の上澄み液の一定量を抽出溶媒で正確に一定倍に希釈したのち,酸性アルミナカートリッジ(例:セップパック アルミナA)に注入し,初めの流出液3mlを捨て,その後の流出液4mlを孔径0.5μm以下のメンブランフィルターでろ過する。
E 定量
試料液10μlをミクロシリンジで高速液体クロマトグラフのカラムに注入し,クロマトグラムのピーク高さまたはピーク面積を測定する。別に標準AM液について同様に操作して測定したピーク高さまたはピーク面積より作成した検量線からAM(C5H6N3Cl)の量を求める。
F 測定条件(例)
1)検出器
紫外部吸光光度計(測定波長295nm)
2)カラム
Finepak SIL C18S(内径4.6mm,長さ150mm,粒径5μm)またはこれと同等のもの(オクタデシル基を化学修飾したシリカゲルを充てん剤として詰めた高速液体クロマトグラフ用カラム)
3)溶離液
高速液体クロマトグラフ用試薬またはこれと同等のメチルアルコール4容と水6容を混合する。
4)流速
1分間1.0mlとする。
6.2 1-アミジノ-2-チオ尿素(ASU)
6.2.1 紫外部吸光光度法
A 適用範囲
1-アミジノ-2-チオ尿素(以下ASUと略す)を含有する肥料に適用する。硝酸塩または有機物を含有する肥料には適用できない場合がある。
B 装置
吸光光度分析装置 紫外部における測定ができる装置であり,吸収セルは石英製とする。
C 試薬液の調製
1)標準ASU液
(C2H6N4S)0.1gを正確にとり,水に溶かして正確に1000mlとする(この液1mlはC2H6N4Sとして0.1mgを含有する)。
2)塩化カリウム緩衝液
6.1.1.C.2)による。
D 試料液の調製
分析試料1gを200ml容の共栓付き三角フラスコに正確にとり,水100mlを正確に加え,マグネチックスターラーで10分間かき混ぜたのち,乾燥ろ紙でろ過する。
E 定量
試料液の一定量(C2H6N4Sとして0.2~1.0mg相当量)を100mlのメスフラスコに正確にとり,塩化カリウム緩衝液2mlを加え,標線まで水を加え,塩化カリウム緩衝液を水で50倍に希釈した空試験の液を対照液として波長262,252及び272nmの吸光度をそれぞれ測定し,各測定値をa,b及びcとし,補正吸光度2a-(b+c)を算出する。別に標準ASU液を数段階(C2H6N4Sとして0.2~1.0mg相当量)に正確にとり,試料液の場合と同一条件で操作して算出した補正吸光度より作成した検量線からASU(C2H6N4S)の量を求める。
6.2.2 高速液体クロマトグラフィー
A 適用範囲
1-アミジノ-2-チオ尿素(以下ASUと略す)を含有する肥料に適用する。
B 装置
紫外部吸光光度分析装置付き高速液体クロマトグラフ
C 試薬液の調製
標準ASU液 6.2.1.C.1)により作成した液を標準ASU原液とする。使用に際してこの原液の一定量を水で正確に40~5倍に希釈する(この液1mlはC2H6N4Sとして2.5~20μgを含有する)。
D 試料液の調製
分析試料1g(C2H6N4Sとして0.25mg相当量以上)を200ml容の三角フラスコに正確にとり,水100mlを正確に加え,マグネチックスターラーで10分間かき混ぜたのち,1分間3000回回転で5分間遠心分離する。上澄み液の一定量を水で正確に一定倍に希釈し,孔径0.5μm以下のメンブランフィルターでろ過する。
E 定量
試料液20μlをミクロシリンジで高速液体クロマトグラフのカラムに注入し,クロマトグラムのピーク高さまたはピーク面積を測定する。別に標準ASU液について同様に操作して測定したピーク高さまたはピーク面積より作成した検量線からASU(C2H6N4S)の量を求める。
F 測定条件(例)
1)検出器
紫外部吸光光度計(測定波長262nm)
2)カラム
μBondapak C18(内径4.6mm,長さ250mm,粒径5μm)またはこれと同等のもの(オクタデシル基を化学修飾したシリカゲル充てん剤として詰めた高速液体クロマトグラフ用カラム)
3)溶離液
1-ヘキサンスルホン酸ナトリウム0.94gをメチルアルコール2容と水8容の混合液に溶かして1000mlとし,酢酸でpH3.15に調製する。試薬はいずれも高速液体クロマトグラフ用またはこれと同等のものを使用する。
4)流速
1分間1.0mlとする。
6.3 4-アミノ-1,2,4-トリアゾール塩酸塩(ATC)
6.3.1 高速液体クロマトグラフィー
A 適用範囲
4-アミノ-1,2,4-トリアゾール塩酸塩(以下ATCと略す)を含有する肥料に適用する。有機物を含有する肥料には適用できない場合がある。
B 装置
紫外部吸光光度分析装置付きの高速液体クロマトグラフ
C 試薬液の調製
標準AT液
4-アミノ-1,2,4-トリアゾール(以下ATと略す)(C2H4N4)0.1gを100mlの褐色メスフラスコに正確にとり,メチルアルコールに溶かし,標線までメチルアルコールを加えて標準AT原液を作成する(この液1mlはC2H4N4として1mg,C2H4N4・HCl(ATC)として1.434mg相当量を含有する)。使用に際してこの原液の一定量をメチルアルコールで正確に5~2倍に希釈する(この液1mlはC2H4N4として0.2~0.5mg,C2H4N4・HClとして0.2867~0.7168mg相当量を含有する)。
D 試料液の調製
分析試料10g(C2H4N4・HClとして28.67mg相当量以上)を250mgのメスフラスコに正確にとり,メチルアルコール100mlを正確に加え,1分間30~40回回転の振り混ぜ機で30分間振り混ぜ,静置後上澄み液の一定量をメチルアルコールで正確に一定倍に希釈し,孔径0.5μm以下のメンブランフィルターでろ過する。
E 定量
試料液10μlをミクロシリンジで高速液体クロマトグラフのカラムに注入し,クロマトグラムのピーク高さまたはピーク面積を測定する。別に標準AT液について同様に操作して測定したピーク高さまたはピーク面積より作成した検量線からATC(C2H4N4・HCl)の量を求める。
F 測定条件(例)
1)検出器
紫外部吸光光度計(測定波長220nm)
2)カラム
Shodex NHpak J-411(内径4.6mm,長さ250mm,粒径5μm)またはこれと同等のもの(アミノ基を化学修飾したシリカゲルを充てん剤として詰めた高速液体クロマトグラフ用カラム)
3)溶離液
アセトニトリル9容とメチルアルコール1容を混合する。試薬はいずれも高速液体クロマトグラフ用またはこれと同等のものを使用する。
4)流速
1分間1.0mlとする。
6.4 N-2,5-ジクロロフェニルスクシナミド酸(DCS)
6.4.1 ナフチルエテレンジアミン法
A 適用範囲
N-2,5-ジクロロフェニルスクシナミド酸(以下DCSと略す)を含有する肥料に適用する。
B 装置
吸光光度分析装置
C 試薬液の調製
1)標準DCS液
DCS(C10H9Cl2NO3)0.5gを正確にとり,メチルアルコールに溶かして正確に1000mlとし,標準DCS原液を作成する(この液1mlはC10H9Cl2NO3として0.5mgを含有する)。使用に際してこの原液の一定量をメチルアルコールで正確に25倍に希釈する(この液1mlはC10H9Cl2NO3として20μgを含有する)。
2)亜硝酸ナトリウム液
亜硝酸ナトリウム(NaNO2)5gを水に溶かして100mlとする。使用に際して作成する。
3)スルファミン酸液
スルファミン酸(NH2SO3H)4.1gを水に溶かして100mlとする。
4)N-(1-ナフチル)エチレンジアミン液
N-(1-ナフチル)エチレンジアミン二塩酸塩[C10N7NH(CH2)2・2HCl]0.25gを水に溶かして25mlとする。使用に際して作成する。
D 試料液の調製
分析試料1~5gを250mlのメスフラスコに正確にとり,メチルアルコール100mlを正確に加え,1分間30~40回回転の振り混ぜ機で1時間振り混ぜたのち,乾燥ろ紙でろ過する。
E 定量
試料液の一定量(C10H9Cl2NO3として0.05~0.6mg相当量)を100mlのメスフラスコに正確にとり,メチルアルコールの量を一定にし,水を加えて約60mlとして混合し,硫酸(5+4)20mlを加えて90~95℃の水浴中で1時間加水分解する[もしこの液が混濁しているならば,冷却後標線まで水を加えて乾燥ろ紙で(5種C)でろ過し,ろ液の一定量(C10H9Cl2NO3として0.05~0.6mg相当量)を100mlのメスフラスコに正確にとり,硫酸(5+4)が20mlとなるようにその不足分を加え,更に水を加えて約80mlとして混合する]。
15℃以下に冷却後亜硝酸ナトリウム液0.5mlを加えてよく振り混ぜ,15分間放置し,更にスルファミン酸液1mlを加えてよく振り混ぜ,20分間放置する。次いでN-(1-ナフチル)エチレンジアミン液0.5mlを加え,更に標線まで水を加えてよく振り混ぜ,30分間放置したのち,空試験の液を対照液として波長530nm付近の吸光度を測定する。同時に標準DCS液を数段階(C10H9Cl2NO3として0.05~0.6mg相当量)に正確にとり,試料液の場合と同一条件で操作して作成した検量線からDCS(C10H9Cl2NO3)の量を求める。
6.4.2 高速液体クロマトグラフィー
A 適用範囲
N-2,5-ジクロロフェニルスクシナミド酸(以下DCSと略す)を含有する肥料に適用する。
B 装置
紫外部吸光光度分析装置付き高速液体クロマトグラフ
C 試薬液の調製
1)標準DCS液
6.4.1.C.1)により標準DCS原液を作成する。使用に際してこの原液の一定量をメチルアルコールで正確に100~25倍に希釈する(この液1mlはC10H9Cl2NO3として5~20μgを含有する)。
2)抽出溶媒
メチルアルコール996容とリン酸4容を混合する。
D 試料液の調製
分析試料1g(C12H9Cl2NO3として0.1mg相当量以上)を250mlのメスフラスコに正確にとり,抽出溶媒100mlを正確に加え,1分間30~40回回転の振り混ぜ機で1時間振り混ぜ,静置後上澄み液の一定量を抽出溶媒で正確に一定倍に希釈し,孔径0.5μm以下のメンブランフィルターでろ過する。
E 定量
試料液10μlをミクロシリンジで高速液体クロマトグラフのカラムに注入し,クロマトグラムのピーク高さまたはピーク面積を測定する。別に標準DCS液について同様に操作して測定したピーク高さまたはピーク面積より作成した検量線からDCS(C10H9Cl2NO3)の量を求める。
F 測定条件(例)
1)検出器
紫外部吸光光度計(測定波長246nm)
2)カラム
μBONDASPHERE5μC18-100Å(内径3.9mm,長さ150mm,粒径5μm)またはこれと同等のもの(オクタデシル基を化学修飾したシリカゲルを充てん剤として詰めた高速液体クロマトグラフ用カラム)
3)溶離液
水をリン酸でpH3に調整した液45容とメチルアルコール55容を混合する。試薬はいずれも高速液体クロマトグラフ用またはこれと同等のものを使用する。
4)流速
1分間0.8mlとする。
6.5 ジシアンジアミド(Dd)
6.5.1 ニトロプルシドナトリウム法
A 適用範囲
ジシアンジアミド(以下Ddと略す)を含有する肥料に適用する。有機物を含有する肥料には適用できない場合がある。
B 装置
吸光光度分析装置
C 試薬液の調製
1)標準Dd液
(C2H4N4)0.4gを正確にとり,水に溶かして正確に100mlとし,標準Dd原液を作成する(この液1mlはC2H4N4として4mgを含有する)。使用に際してこの原液の一定量を水で正確に10倍に希釈する(この液1mlはC2H4N4として0.4mgを含有する)。
2)ニトロプルシドナトリウム液
ニトロプルシドナトリウム(ペンタシアノニトロシル鉄(Ⅲ)酸ナトリウム二水和物)[Na2Fe(CN)5NO・2H2O]5.96gを水に溶かして100mlとする。
3)フェリシアン化カリウム液 フェリシアン化カリウム[K3Fe(CN)6]6.59gを水に溶かして100mlとする。
4)発色試薬液
使用に際してニトロプルシドナトリウム液10mlを50mlのメスフラスコに正確にとり,フェリシアン化カリウム液10ml及び10%水酸化ナトリウム液5mlを正確に加え,更に標線まで水を加えて振り混ぜ,30分間放置する。
5)イオン交換樹脂
粒径125~212μmの強酸性陽イオン交換樹脂(例:アンバーライトIR120)をビーカーにとり,塩酸(1+4)を加えて洗浄し,その洗液を捨て,更に12%塩化ナトリウム液を加えて洗浄し,その洗液を捨てる。この操作を数回繰り返し,再び塩酸(1+4)で同様の操作を行って樹脂をH+型に変え,水で洗浄する。
D 試料液の調製
a 尿素を含有しない場合
分析試料10gを250mlのメスフラスコに正確にとり,アセトン200mlを正確に加え,1分間30~40回回転の振り混ぜ機で1時間振り混ぜたのち,乾燥ろ紙でろ過する。ろ液の一定量をビーカーに正確にとり,70~75℃の水浴上で蒸発乾固する。残留物を少量の水に溶かして50mlのメスフラスコに水でことごとく移し,標線まで水を加える。
b 尿素を含有する場合
液だめ及びガラスろ過器付きクロマト管(内径7.7mm,長さ150mm)に水に懸濁させたイオン交換樹脂を高さ150mmまで流し込み,液面がイオン交換樹脂の上面から3mmの高さになるまで水を流出させ,更に水を加え,流出液が中性になるまで同様に流出させてカラムを調製する。
前記aの試料液1ml(C2H4N4として1~5mg相当量)を調製されたカラムに正確に注ぎ,1分間に1mlの速度で液面がイオン交換樹脂の上面から3mmの高さになるまで流出させたのち,水を少量ずつ加えて同じ速度で50mlのメスフラスコに25mlまで溶出させる。
(付記)
1. 分析試料に油が共存する場合には,前記aの残留物にジエチルエーテルを加えて油を溶かし,デカンテーションによりこれを除去したのち,「残留物を少量の水に溶かして」以下により試料液を作成する。
2. 使用後のカラムは,カラムに塩酸(1+4)20~30mlを加え,液面がイオン交換樹脂の上面から3mmの高さになるまで流出させ,更に水を加え,流出液が中性になるまで同様に流出させて洗浄し,再調製することができる。
E 定量
前記aでは試料液の一定量(C2H4N4として1~5mg相当量)を50mlのメスフラスコに正確にとり,水を加えて25mlとし,前記bではカラムクロマトグラフィー操作後の溶出液に,それぞれ発色試薬液3mlを加え,更に標線まで水を加えて振り混ぜ,1時間放置したのち,空試験の液を対照液として波長515nm付近の吸光度を測定する。同時に標準Dd液を数段階(C2H4NO4として1~5mg相当量)に正確にとり,試料液の場合と同一条件で操作して作成した検量線からDd(C2H4N4)の量を求める。
6.5.2 高速液体クロマトグラフィー
A 適用範囲
ジシアンジアミド(以下Ddと略す)を含有する肥料に適用する。
B 装置
紫外部吸光光度分析装置付き高速液体クロマトグラフ
C 試薬液の調製
標準Dd液
Dd(C2H4N4)1gを正確にとり,メチルアルコールに溶かして正確に1000mlとし,標準Dd原液を作成する(この液1mlはC2H4N4として1mgを含有する)。使用に際してこの原液の一定量をメチルアルコールで正確に1000~20倍に希釈する(この液1mlはC2H4N4として1~50μgを含有する)。
D 試料液の調製
分析試料1g(C2H4N4として0.1mg相当量以上)を200ml容の分液漏斗に正確にとり,メチルアルコール100mlを正確に加え,10分間激しく振り混ぜ,静置後上澄み液の一定量をメチルアルコールで正確に一定倍に希釈し,孔径0.5μm以下のメンブランフィルターでろ過する。
(付記)
1.有機物を含有する肥料の場合には,上記のメチルアルコールで正確に一定倍に希釈した液の適量を,クロマト管(内径10mm,長さ250mm)に粒径75~150μmのカラムクロマトグラフ用シリカゲル(例:ワコーゲルC-200;110℃で2時間乾燥したもの)2.5g及び無水硫酸ナトリウム10mm(高さ)を順次乾式充てんしたカラムに注ぎ,初めの流出液数mlを捨て,その後の流出液を孔径0.5μm以下のメンブランフィルターでろ過して試料液とする。
2.有機物を含有し,かつ水分の多い肥料の場合には,前記の上澄み液の一定量を三角フラスコにとり,無水硫酸ナトリウムを加えて脱水し,乾燥ろ紙でろ過したのち,上記の(付記)1.の操作を行う。
E 定量
試料液10μlをミクロシリンジで高速液体クロマトグラフのカラムに注入し,クロマトグラムのピーク高さまたはピーク面積を測定する。別に標準Dd液について同様に操作して測定したピーク高さまたはピーク面積より作成した検量線からDd(C2H4N4)の量を求める。
F 測定条件(例)
1)検出器
紫外部吸光光度計(測定波長215nm)
2)カラム
UNISILQNH2(内径4.6mm,長さ250mm,粒径5μm)またはこれと同等のもの(アミノ基を化学修飾したシリカゲルを充てん剤として詰めた高速液体クロマトグラフ用カラム)
3)溶離液
アセトニトリル6容とメチルアルコール1容を混合する。試薬はいずれも高速液体クロマトグラフ用またはこれと同等のものを使用する。
4)流速
1分間0.5mlとする。
6.6 2-スルファニルアミドチアゾール(ST)
6.6.1 紫外部吸光光度法
A 適用範囲
2-スルファニルアミドチアゾール(以下STと略す)を含有する肥料に適用する。硝酸塩または有機物を含有する肥料には適用できない場合がある。
B 装置
吸光光度分析装置 紫外部における測定ができる装置であり,吸収セルは石英製とする。
C 試薬液の調製
1)標準ST液
ST(C9H9N3O2S2)0.1gを正確にとり,水に溶かして正確に1000mlとする(この液1mlはC9H9N3O2S2として0.1mgを含有する)。
2)塩化カリウム緩衝液
6.1.1.C.2)による。
D 試料液の調製
6.1.1.Dによる。
E 定量
試料液の一定量(C9H9N3O2S2として0.5~2.0mg相当量)を100mlのメスフラスコに正確にとり,塩化カリウム緩衝液20mlを加え,標線まで水を加え,塩化カリウム緩衝液を水で5倍に希釈した空試験の液を対照液として波長280.5,270.5及び290.5nmの吸光度をそれぞれ測定し,各測定値をa,b及びcとし,補正吸光度2a-(b+c)を算出する。別に標準ST液を数段階(C9H9N3O2S2として0.5~2.0mg相当量)に正確にとり,試料液の場合と同一条件で操作して算出した補正吸光度より作成した検量線からST(C9H9N3O2S2)の量を求める。
6.6.2 高速クロマトグラフィー
A 適用範囲
2-スルファニルアミドチアゾ-ル(以下STと略す)を含有する肥料に適用する。
B 装置
紫外部吸光光度分析装置付き高速液体クロマトグラフ
C 試薬液の調製
1)標準ST液
6.6.1.C.1)により作成した液を標準ST原液とする。使用に際してこの原液の一定量を抽出溶媒で正確に50~12.5倍に希釈する(この液1mlはC9H9N3O2S2として2~8μgを含有する)。
2)抽出溶媒
6.1.2.C.2)による。
D 試料液の調製
分析試料1g(C9H9N3O2S2として0.2mg相当量以上)を200ml容の共栓付き三角フラスコに正確にとり,抽出溶媒100mlを正確に加え,マグネチックスターラーで15分間かき混ぜたのち,1分間3000回回転で5分間遠心分離する。上澄み液の一定量を抽出溶媒で正確に一定倍に希釈し,孔径0.5μm以下のメンブランフィルターでろ過する。
(付記)
有機物を含有する肥料の場合には,6.1.2.D.(付記)による。
E 定量
試料液10μlをミクロシリンジで高速液体クロマトグラフのカラムに注入し,クロマトグラムのピーク高さまたはピーク面積を測定する。別に標準ST液について同様に操作して測定したピーク高さまたはピーク面積より作成した検量線からST(C9H9N3O2S2)の量を求める。
F 測定条件(例)
1)検出器
紫外部吸光光度計(測定波長285nm)
2)カラム
6.1.2.F.2)による。
3)溶離液
高速液体クロマトグラフ用試薬またはこれと同等のメチルアルコール2容と水8容を混合する。
4)流速
1分間1.0mlとする。
6.7 チオ尿素(TU)
6.7.1 ニトロプルシドナトリウム法
A 適用範囲
チオ尿素(以下TUと略す)を含有する肥料に適用する。
B 装置
1)吸光光度分析装置
2)イオン交換装置
内径0.5cm,高さ15cmのガラス管の下端にガラス綿を敷いて強塩基性樹脂(アンバーライトIRA400など)を高さ約10cm(交換容量約5meq)に詰めたものを用い,定量の都度12%塩化ナトリウム液(または8%水酸化ナトリウム液)約50mlと水50~100mlとを,チオシアン酸及び塩化物(またはアルカリ)の反応がなくなるまで,交互に1分間2~4mlの流速で通してカラムを作成する。樹脂はあらかじめビーカー中で塩酸(1+5),水酸化ナトリウム液,水の順に数回繰り返して洗浄し,コンディショニングする。
C 試薬液の調製
1)標準TU液
特級TU[(NH2)2CS](硫酸デシケーター中で24時間以上乾燥したもの)5gを1000mlのメスフラスコに正確にとり,水に溶かして更に標線まで水を加える。この液の一定量を水で正確に100倍に希釈する[この液1mlは(NH2)2CSとして50μgを含有する]。
2)塩化鉄(Ⅲ)液
塩化鉄(Ⅲ)(FeCl3・6H2O)5gを塩酸(1+5)100mlに溶かして着色瓶に貯蔵する。
3)ニトロプルシドナトリウム液
3%ニトロプルシドナトリウム(ペンタシアノニトロシル鉄(Ⅲ)酸ナトリウム二水和物)[Na2Fe(CN)6NO・2H2O]液と3%水酸化ナトリウム液とを等量混合して25℃に5時間放置したのち,着色瓶に入れて冷暗所に貯蔵し,使用に際してろ過する。ただしこの液は長時間の保存に耐えない。
4)酢酸ナトリウム緩衝液
酢酸ナトリウム(NaCH3・CO2・3H2O)27.2gを水に溶かし塩酸(1+10)約170mlを加え,更に水を加えて1000mlとしpHを3.3に調製する。
D 試料液の調製
分析試料2.5gを小型乳鉢に正確にとり,少量のアセトンを加えてよくすりつぶし,その上澄み液を250mlのメスフラスコに入れ,更にこの操作を3回繰り返したのち乳鉢内の不溶解物をことごとくメスフラスコに移し,アセトンを加えて約200mlとし1分間30~40回回転の振り混ぜ機で30分間振り混ぜたのち,標線までアセトンを加えて乾燥ろ紙でろ過する。
E 定量
定量に先だち分析試料の約1%水溶液に塩化鉄(Ⅲ)液を加えてチオシアン酸塩の有無を調べる。
a チオシアン酸塩を含有しない場合
試料液の一定量[(NH2)2CSとして約5mgがよい]をビーカーに正確にとり,70~75℃の水浴中で蒸発乾固したのち,残留物を水に溶かして正確に100mlとし,必要があれば乾燥ろ紙でろ過する。この液の一定量(吸光度0.1~0.4の範囲がよい)を100mlのメスフラスコに正確にとり,適量の水及びニトロプルシドナトリウム液5mlを加えて振り混ぜたのち,酢酸ナトリウム緩衝液20mlを加え更に標線まで水を加える。30~60分後空試験の液を対照液として波長600nm付近の吸光度を測定する。同時に標準TU液を数段階に正確にとり,試料液の場合と同一条件で操作して作成した検量線からTU[(NH2)2CS]の量を求める。
b チオシアン酸塩を含有する場合
試料液の一定量[(NH2)2CSとして約5mgがよい]をビーカーに正確にとり,70~75℃の水浴中で蒸発乾固したのち,残留物は少量の水(25ml以内)に溶かし,イオン交換装置に移して1分間約2mlの流速でカラムを通し,更に同一流速で水を通してその流出液100mlを正確にとる。流出液の一定量(吸光度0.1~0.4の範囲がよい)を100mlのメスフラスコに正確にとり,前項aの「適量の水」以下によりTUを定量する。
7 参考
7.1 二クロム酸酸化による有機炭素の定量法
A 適用範囲
汚泥肥料,たい肥等の有機肥料の有機炭素あるいは炭素-窒素比を求めるのに適する。本法は炭酸塩が共存しても誤差とならない簡便なよい方法であるが,有機物は炭素の平均酸化数が零となるような化学組成を有するという仮定に基づいているので,実際には多少の誤差が避けられないし,また塩化物や2価鉄の共存により正の誤差も生ずる。
B 試薬液の調製
1)標準二クロム酸カリウム液
標準試薬二クロム酸カリウム(K2Cr2O7)(めのう乳鉢を用いて粉末とし,100~110℃に3~4時間保ったのち,硫酸デシケーター中で放冷したもの)9.806gを水に溶かして正確に1000mlとし,0.2M(1/6K2Cr2O7)溶液を作成する。
2)標準硫酸第一鉄アンモニウム液
硫酸第一鉄アンモニウム[FeSO4・(NH4)2SO4・6H2O]80gを,硫酸20mlを含む水1000mlに溶かし,約0.2M溶液を作成する。使用日ごとにフェニルアントラニル酸液を指示薬として標準二クロム酸カリウム液を滴定して補正係数(F)を定める。
3)二クロム酸カリウム-硫酸液
二クロム酸カリウム(K2Cr2O7)40gを水1000mlに溶かし,冷却しながら硫酸1000mlを少しずつ混合しながら加える。
4)指示薬
フェニルアントラニル酸液 N-フェニルアントラニル酸(C13H11O2N)0.2gと無水炭酸ナトリウム(Na2CO3)0.2gを水5mlに溶かし,水を加えて100mlとする。
C 定量
分析試料5g以上をめのう乳鉢でよく摩砕したものの一定量(Cとして40~60mg相当量,Wmg)を100ml容の三角フラスコに正確にとり,二クロム酸カリウム-硫酸液10mlを正確に加える。別に空試験として二クロム酸カリウム-硫酸液だけを正確に10ml入れた100ml容の三角フラスコを用意する。
これらの三角フラスコを熱板状に置き,図3に示す凝縮器を差す。熱板が約200℃に昇温し,沸騰が始まってから30分後に加熱を止める。2~3分間放置後凝縮器を抜いて三角フラスコを熱板から降ろす。放冷後水約10mlを加える。
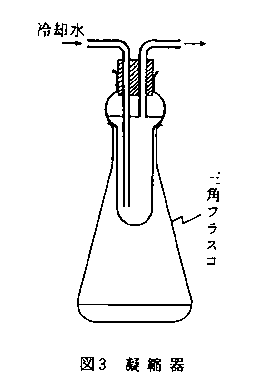
次に加熱分解後の二クロム酸の残量を標準硫酸第一鉄アンモニウム液で滴定する。この際指示薬フェニルアントラニル酸液は0.25mlまたはそれに相当する滴数を二クロム酸イオンの色がごく薄くなった時に加える。滴定の終点は暗赤紫色から鮮明な緑色に変わった時である。滴定値(Tml)は空試験値(Bml)の1/2~3/4が適当である(この範囲を外れるときは分析試料の採取量を変えて操作をやり直す)。次式により有機炭素(C)の量を算出する。
C(%)=(B-T)×F×60/W
7.2 クエン酸二アンモニウム液可溶性リン(リン酸)の定量法
A 適用範囲
可溶性(クエン酸塩液可溶性)リン(リン酸)は4.2.1.C.cにより試料液を作成するのであるが,長時間を要する欠点がある。本法は2%クエン酸二アンモニウム液により浸出して試料液とするものであり,多くの複合肥料などにおいて可溶性リン(リン酸)と相似した値をもたらす。しかし,魚かす,骨粉などを原料とした複合肥料や過リン酸石灰などでは可溶性リン(リン酸)との差が大きい場合があるので,適用に際しては予備試験を行い,かたよりを避けるなどの配慮が必要である。
B 試薬液の調製
1)クエン酸二アンモニウム液
クエン酸二アンモニウム[(NH4)2HC6H5O7]20gを水に溶かして1000mlとする。
2)その他
4.2各項に記載の試薬液を作成する。
C 試料液の調製
分析試料1gを250mlのメスフラスコに正確にとり,クエン酸二アンモニウム液150mlを加え,密栓をして振り混ぜ,65℃の水浴中で15分ごとに振り混ぜながら1時間作用させたのち,水を加えて冷却し,更に標線まで水を加えて直ちに乾燥ろ紙でろ過する。
D 定量
試料液の一定量を正確にとり,4.2によりリン(P)またはリン酸(P2O5)の量を求める。
(付記)
7.3 中性クエン酸塩液可溶性リン(リン酸)の定量法
A 適用範囲
本法はアメリカ分析化学者協会(AOAC)法に準拠した方法である。リン(リン酸)の浸出剤に中性のクエン酸アンモニウム液を用いるため,アルカリ性である4.2.1.B.3)のペーテルマンクエン酸塩液による定量値とは一致しない。また,この中性クエン酸塩液可溶性リン(リン酸)には水溶性リン(リン酸)を含めないので,この点も4.2.1.C.cの可溶性(クエン酸塩液可溶性)リン(リン酸)とは異なる。
B 試薬液の調製
1)中性クエン酸アンモニウム液
クエン酸370gを水1500mlに溶かし,アンモニア水345mlを加えてほぼ中和する。冷却後3%クエン酸液またはアンモニア水(1+7)でpH7.0に調整する。必要があれば20℃で比重1.09の溶液となるように水で希釈する。使用に際してpH7.0に再調整する。
2)その他
4.2各項に記載の試薬液を作成する。
C 試料液の調製
1)水溶性リン(リン酸)を含有する場合
分析試料1gをろ紙上に正確にとり,少量ずつの水でろ液が約250mlとなるまで洗浄したのち,あらかじめ65℃に加熱した中性クエン酸アンモニウム液100mlを入れたフラスコにろ紙及び残留物を1時間以内に移す。栓をして,ろ液が完全に崩壊するまで振り混ぜ,65℃の水浴中で時々振り混ぜながら1時間作用させたのち,直ちにろ紙(3種)を用いて速やかに吸引ろ過し,65℃の水でろ液が約350mlとなるまで洗浄する。次にⅰ)またはⅱ)により試料液を作成する。
ⅰ)ろ紙及び残留物を乾燥後白金るつぼに移し,強熱したのち塩酸10~15mlに溶かし,放冷後水を加えて正確に250mlとし,乾燥ろ紙でろ過する。
ⅱ)ろ紙及び残留物をトールビーカーに移し,硝酸20~30mlを加えて30~45分間穏やかに煮沸する。放冷後過塩素酸10~20mlを加え加熱・蒸発し,過塩素酸の白煙が発生するようになったならば時計皿で覆い,溶液が澄明になるまで乾固させないように注意しながら静かに加熱する。暫時放冷後水50mlを加え数分間煮沸したのち放冷し,水を加えて正確に250mlとし乾燥ろ紙でろ過する。
2)水溶性リン(リン酸)を含有しない場合
分析試料1gをあらかじめ65℃に加熱した中性クエン酸アンモニウム液100mlを入れたフラスコにとり,前記1)の「65℃の水浴中で」以下に従い試料液を作成する。
D 定量
試料液の一定量につき,4.2.1.D,4.2.2.Dまたは4.2.3.E.aによりリン(リン酸)を定量し,中性クエン酸塩液不溶性リン(リン酸)の量を求める。別に4.2によりリン(リン酸)全量及び水溶性リン(リン酸)を定量し,次式により中性クエン酸塩液可溶性リン(リン酸)の量を算出する。
中性クエン酸塩液可溶性P(P2O5)(%)=P(P2O5)全量(%)-水溶性P(P2O5)(%)-中性クエン酸塩液不溶性P(P2O5)(%)
7.4 酢酸塩緩衝液可溶性ケイ素(ケイ酸)の定量法
A 適用範囲
ケイ酸質肥料の有効ケイ素(ケイ酸)は可溶性(0.5M塩酸可溶性)ケイ素(ケイ酸)で評価するのであるが,スラグの種類により肥効発現の遅速を十分に示すことができない場合がある。本法はこのような肥効発現の速度を示す一つの尺度として提案するものであるが,この方法によってもなお肥効を完全に評価することはできない場合もあることに留意しなければならない。
B 試薬液の調製
1)酢酸塩緩衝液
酢酸58mlを水に加えて約500mlとする。ガラス電極pH計を用い10%水酸化ナトリウム液を加えてpH4.0とし,更に水を加えて1000mlとする。
C 試料液の調製
分析試料1gを250mlのメスフラスコに正確にとり,30℃の酢酸塩緩衝液150mlを加え1分間30~40回回転の振り混ぜ機で1時間振り混ぜたのち(浸出中は30℃に保つこと),標線まで水を加えて直ちに乾燥ろ紙でろ過する。
D 定量
作成直後の試料液につき,4.4.3.Dにより定量する。
(付記)
4.4.3.Dの代わりに,試料液の一定量(25ml以下)につき,硝酸を加えて予備的分解したのち4.4.2.Cにより定量することもできる。ただし,過塩素酸の加熱時には,爆発の危険性があるので乾固させないよう十分注意しなければならない。
7.5 誘導結合プラズマ(ICP)発光分光法による各種元素の定量法
A 適用範囲
誘導結合プラズマ(ICP)の高温を利用した発光分光分析法により,リン,ケイ素,ホウ素などを含む多くの元素を定量することができる。溶液試料を用いるので,各項に記載された試料液を適宜希釈することによって,多くの場合定量が可能である。
B 装置
発光分光分析装置 ICP光源
C 試薬液の調製
前章までの各項に記載されて標準原液を使用に際して所定濃度に正確に希釈する。この際には試料液に合わせて使用する酸の種類,濃度などを一定にする。
D 試料液の調製
前章までの各項による。
E 定量
試料液を必要により所定濃度に正確に希釈し(この際,酸の種類・濃度を,例えば,塩酸0.1~0.5Mのように,一定にする),この液を発光分光分析装置に吸入噴霧させて発光強度を測定する。同時に標準液について同一条件で操作して作成した検量線から各元素の量を求める。
(付記)
1.測定できる元素は多くのものがあるが,その一例をあげると,アルミニウム,ホウ素,カルシウム,カドミウム,コバルト,クロム,鉄,マグネシウム,マンガン,モリブデン,ニッケル,リン,鉛,ケイ素,亜鉛などがある。測定濃度は元素,測定波長,装置などにより異なるので,あらかじめ最適濃度範囲を決めておく必要がある。
2.共存物質の影響については,多量元素の測定ではあまり問題とならないが,微量元素の測定の場合にはスペクトル干渉,バックグラウンド干渉があるので,あらかじめそれらの影響の大きさを調べて補正することが必要である。
7.6 イオンクロマトグラフィーによる陰イオンの定量法
A 適用範囲
水溶液中の陰イオンの多くは本法により分離され,個々に定量することができる。
B 装置
イオンクロマトグラフ
C 試薬液の調製
1)標準陰イオン液 標準フッ素原液[5.26.1.C.1)による],標準塩素原液(塩化ナトリウム1.648gを水に溶かして正確に1000mlとし,Clとして1000μg/mlの溶液とする),標準リン原液[4.2.3.C.1)による],標準硝酸塩原液[4.1.3.3.C.1)による],標準硫酸塩原液(硫酸カリウム5.434gを水に溶かして正確に1000mlとし,SO4-Sとして1000μg/mlの溶液とする)の各所定量を同一のメスフラスコに正確にとり,標線まで水を加える。
2)溶離液
炭酸水素ナトリウム0.252g及び炭酸ナトリウム0.254gを水に溶かして1000mlとする。
D 試料液の調製
E 定量
試料液を適当な濃度にまで水で正確に希釈し,イオンクロマトグラフに注入し,電気伝導率検出器の指示を記録する。各陰イオンについてピーク高さ(またはピーク面積)を求める。別に標準陰イオン液について測定したピーク高さ(またはピーク面積)から作成した検量線により陰イオンの量を求める。
(付記)
1.溶離液は装置により異なることがある。
2.上記以外の陰イオンとして亜硝酸,臭化物なども同時に測定できる。




